Мышечная дистрофия: причины, симптомы и лечение заболеваний мышц. Прогрессирующая мышечная дистрофия лечение
Прогрессирующая мышечная дистрофия - лечение и симптомы
Прогрессирующая мышечная дистрофия – это недуг, который при неправильном лечении или при его отсутствии, может привести к ограничению жизнедеятельности человека. Дело в том, что атрофия мышц уменьшает объем и силу движений, является причиной паралича и пареза. Существуют различные виды мышечной дистрофии, которые могут захватывать мышцы лица, конечности плечевого или тазового пояса. Дистрофия мышц считается типом атрофии, медленно прогрессирующим заболеванием. В нашей статье мы расскажем вам про лечение и симптомы прогрессирующей мышечной дистрофии.
Лечение прогрессирующей мышечной дистрофии
Лечение симптомов прогрессирующей мышечной дистрофии
1.
Прежде всего, больной с симптомами прогрессирующей мышечной дистрофии должен сам заниматься своим лечением до тех пор пока это будет в его силах он должен делать физические упражнения. Хорошо помогает плавание в бассейне. Отлично помогают в лечении прогрессирующей мышечной дистрофии соляные озёра, и море. Вода содержит микроэлементы и поддерживает организм при лечении.
2.
Хорошо при прогрессирующей мышечной дистрофии помогают профессионалы в области рефлексотерапии. В частности грамотные методы акупунктуры могут провоцировать регенеративные процессы, что позволяет спасать мышцы от их деградации и вырождения при лечении.
3.
Ещё один метод рефлексотерапии при лечении симптомов прогрессирующей мышечной дистрофии это использование температур на определённых точках тела. Это также показывает себя с хорошей стороны в ходе лечения прогрессирующей мышечной дистрофии.
4.
Безусловно, и традиционный массаж может помочь поддержать быстро слабеющую трофику тела при лечении прогрессирующей мышечной дистрофии.
5.
Физиотерапии, особенно с использованием электрических разрядов могут провоцировать тонус мышц, и иногда запускают обратную реакцию, хотя случается это на ранних стадиях симптомов прогрессирующей мышечной дистрофии.
6.
Активно для лечения прогрессирующей мышечной дистрофии начали применять медикаментозные методики. Препараты направлены на стимуляцию роста мышечной массы, иногда они помогают сократить симптомы прогрессирующего процесса.
7.
Кроме того, больного с симптомами прогрессирующей мышечной дистрофией сильно поддерживают в плане витаминов и необходимых микроэлементов во время лечения. Если вдруг такому больному организму не будет чего-то хватать, то это халатность. Это наплевательское отношение к шансу. Поэтому ввод витаминов не прекращают.
8.
Также при симптомах прогрессирующей мышечной дистрофии используют для лечения стимулирующие препараты. Больной испытывает усталость, ему хочется лежать, отдыхать. Чтобы подавить эту меланхолию используют стимуляторы. Это могут быть привычный женьшень, радиола розовая, а могут быть более серьёзные препараты.
Лечение прогрессирующей мышечной дистрофии Дюшена и ее симптомов
1.
Основное лечение при прогрессирующей мышечной дистрофии Дюшена направлено на поддержание мышечной активности больного. С ребёнком занимаются спортом для лечения симптомов прогрессирующей мышечной дистрофии. Физические нагрузки, аэробика, плавание, всё это помогает сократить скорость деградации и вырождения тканей. Если ввести больного в режим постоянной работы, то это приносит отличные результаты.
2.
Проблема в том, что заставить двухлетнего ребёнка преодолевая немощь, прыгать, сложно. Поэтому используют массу физиопроцедур для лечения. Это воздействие токами, магнитными установками, тепловыми и холодовыми процедурами.
3.
Особое место в лечении прогрессирующей мышечной дистрофии Дюшена отводится массажу и комплексу рефлексотерапии. Эти средства, при разумном использовании помогут долго поддерживать организм больного в тонусе, но это опять же труд и усилия больного и врачей.
Лечение медленно прогрессирующей мышечной дистрофии
1.
На первых этапах развития симптомов мышечной дистрофии необходимо пройти электромиографию. Причем, в обследовании должны принимать участие также и члены семьи больного. Это позволяет выявить наследственные патологии и спрогнозировать интенсивность развития недуга, чтобы знать, каким образом его впоследствии лечить.
2.
Как такового специфического лечения мышечной дистрофии нет. Но пациенту назначают для лечения массаж, витаминотерапию, электростимуляцию и прочие подобные процедуры.
3.
Важно вести активный образ жизни на уровне тех возможностей, которые имеются у человека. Позитивное влияние на состояние мышц при симптомах прогрессирующей мышечной дистрофии оказывает лечебная физкультура.
4.
Тело ежедневно можно протирать смоченной в воде салфеткой для лечения. Начинать следует с груди, а затем переходить к спине и конечностям. Процедуру проводят в течение 2 минут, затем укутываются в простыню и до высыхания воды находятся в теплом помещении. Это позволяет улучшить тонус мышц.
5.
Вышеуказанные обтирания при симптомах прогрессирующей мышечной дистрофии можно проводить для лечения с использованием яблочного уксуса или смешать его с водой в соотношении 1:1.
6.
В комплексе с медикаментозным лечением дистрофии мышц, которое назначил врач, знающий, как лечить мышечную дистрофию эффективно, можно использовать рецепты народной медицины. Например, 1/3 банки следует заполнить измельченным корнем дягиля, а остальные 2/3 – водкой. Такую настойку выдерживают 9 дней, в темном месте, периодически встряхивая. По истечении указанного срока настойку процеживают, и полученное средство лечения втирают в руки, ноги, спину и грудь перед сном, предварительно обтерев водой тело и разогрев его. После процедуры растирания важно лечь под одеяло, чтобы тело прогрелось. Подобную процедуру нужно выполнять ежедневно в течение длительного времени при симптомах прогрессирующей мышечной дистрофии для лечения.
7.
1-2 раза в неделю при мышечной дистрофии для лечения можно принимать ванны с английской солью (можно также использовать обыкновенную или морскую соль). На 100 литров воды требуется 3-5 килограмма соли.
8.
Позитивное влияние на состояние мышц при симптомах прогрессирующей мышечной дистрофии оказывают также ванночки для ног с использованием отваров сенной трухи, овсяной соломы, хвойных веток, березовых листьев или корня лопуха. Можно использовать контрастные ванночки с водой комнатной температуры и более горячей водой (50-55 градусов).
При грамотном подходе к лечению мышечной дистрофии развитие заболевания удается замедлить, обеспечив трудоспособность на долгие годы!
Симптомы прогрессирующей мышечной дистрофии
Какими симптомами проявляется прогрессирующая мышечная дистрофия?
1.
Начало прогрессирующей мышечной дистрофии знаменуют симптомы: мышечная слабость в ногах и тазовых связках. При симптомах прогрессирующей мышечной дистрофии Дюшена малыш быстро устаёт, начинает косолапить, хромать, предпочитает поднять ноги выше уровня тела.
2.
В дальнейшем развитии, мышцы полностью слабеют и ребёнок лишается опорной функции, это основной симптом прогрессирующей мышечной дистрофии.
3.
Далее мышечная дистрофия Дюшена поражает руки, и ребёнок постепенно, медленно, а иногда очень быстро теряет контроль над ними.
4.
Далее симптомы прогрессирующей мышечной следует шея, плечевой пояс мышц и спина. Наступает почти полный паралич при симптомах прогрессирующей мышечной дистрофии.
5.
Последними из внешнего мышечного скелета отказывают лицевые мышцы.
Дальнейшая деградация мышечной ткани при симптомах прогрессирующей мышечной дистрофии поражает лёгкие, сосуды, иногда сердечную мышцу. Полностью лечению прогрессирующая мышечная дистрофия Дюшена не поддается. Однако есть возможность продлить работоспособный период жизни больного с симптомами прогрессирующей мышечной дистрофии.
www.medmoon.ru
Прогрессирующие мышечные дистрофии. Лечение
Термином мышечные дистрофии называется группа генетически детерминированных расстройств, характеризующихся прогрессирующими дегенеративными изменениями в мышечных волокнах без первичной патологии периферического (нижнего) мотонейрона.
Различные формы отличаются друг от друга типами наследования, сроками начала процесса, характером и быстротой его течения, своеобразием топографии мышечных атрофии, наличием или отсутствием псевдогипертрофий и сухожильных ретракций и другими признаками.
Большинство мышечных дистрофий хорошо изучено клинически, их подробное описание сделано еще в конце прошлого века. Но, несмотря на почти вековую историю изучения миодистрофий, вопросы их патогенеза и лечения остаются до сего времени неразрешенными. Большие надежды возлагаются на молекулярную генетику, с помощью которой определено местонахождение генов уже многих нозологических форм.
Диагностика мышечных дистрофий нередко представляет большие трудности. Имеется большая вариабельность клинических проявлений, а малое число членов семьи затрудняет определение типа наследования.
Характерным моторным дефектом у больных с мышечными дистрофиями является “утиная” походка: больной ходит переваливаясь к боку на бок. Она связана главным образом со слабостью ягодичных мышц, прежде всего средней и малой, которые фиксируют таз относительно бедренной кости. В результате при заболевании возникает наклон таза в сторону неопорной ноги (феномен Тренделенбурга) и компенсаторный наклон туловища в противоположную сторону (феномен Дюшенна). При ходьбе сторона наклона постоянно меняется. Указанные изменения можно проверить и в пробе Тренделенбурга, попросив больного поднять одну ногу, согнув ее под прямым углом в коленном и тазобедренном суставах: таз на стороне поднятой ноги опускается (а не поднимается как в норме) из-за слабости средней ягодичной мышцы опорной ноги.
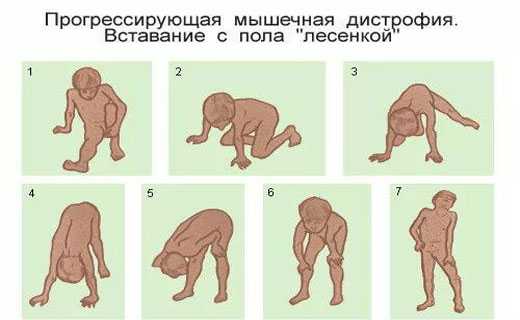
Поднимаясь из горизонтального положения, больной с выраженной мышечной слабостью проксимальных мышц с трудом переворачивается на живот, затем, упираясь руками в пол, становится на четвереньки и после этого, упираясь руками в голени, затем в бедра, постепенно выпрямляется. Этот феномен “набирания по себе” носит название приема Говерса. Часто он связан со слабостью больших ягодичных мышц.
Миодистрофия Дюшенна
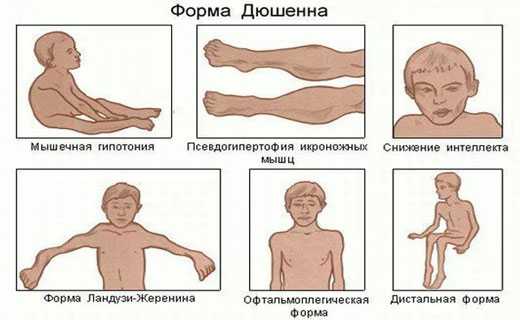
Псевдогипертрофическая мышечная дистрофия Дюшенна встречается чаще всех других заболеваний мышечной системы (30 на 100000 живых новорожденных). Характеризуется ранним началом и злокачественным течением. Классическая картина проявляется изменением походки у ребенка в возрасте 2-5 лет, к 8-10 годам дети ходят уже с трудом, к 14-15 годам они, как правило, полностью обездвижены. У детей более раннего возраста начальные симптомы проявляются отставанием двигательного развития: они позднее начинают ходить, не умеют бегать и прыгать. Больные умирают на 2-3-м десятилетии жизни.
Одними из первых признаков заболевания являются уплотнения икроножных мышц и постепенное увеличение их объема за счет псевдогипертрофий. Атрофии мышц бедра, тазового пояса нередко маскируются хорошо развитой подкожной жировой клетчаткой. Постепенно процесс принимает восходящее направление и распространяется за плечевой пояс, мышцы спины, а затем и на проксимальные отделы рук.
Миодистрофия Беккера. Наряду с тяжелой, злокачественной формой Х-сцепленной миодистрофией Дюшенна существует доброкачественная форма - болезнь Беккера. По клиническим симптомам она очень напоминает форму Дюшенна, однако начинается, как правило, позднее - в 10-15 лет, течет мягко, больные длительно сохраняют работоспособность, в возрасте 20- 30 лет и позже еще могут ходить. Фертильность не снижена, поэтому заболевание иногда прослеживается в нескольких поколениях семьи: больной мужчина через свою дочь передает заболевание внуку (“эффект деда”). Начальные симптомы, как и при болезни Дюшенна, проявляются слабостью в мышцах тазового пояса, затем в проксимальных отделах нижних конечностей. У больных изменяется походка, они испытывают затруднение при подъеме по лестнице, при вставании с низкого сиденья. Характерны псевдогипертрофии икроножных мышц. Ретракция пяточных (ахилловых) сухожилий выражена менее резко, чем при болезни Дюшенна.
Миодистрофия Ландузи-Дежерина. Заболевание передается по аутосомно-доминантному типу с высокой пенетрантностью, но несколько вариабельной экспрессивностью. Встречается гораздо реже, чем миодистрофия Дюшенна (0,4 на 100 тыс. населения). Предполагают, что ген этого заболевания локализован в 4-й хромосоме. Женщины болеют чаще мужчин (3:1), Физические перегрузки, интенсивные занятия спортом, а также нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни.
Миодистрофия Ландузи-Дежерина - сравнительно благоприятно текущая форма мышечной патологии. Начинается она еще в возрасте около 20 лет, иногда позже. Однако в семейных случаях заболевания, когда можно проследить за младшими членами семьи в динамике, удается выявить некоторую слабость мышц, например мышц лица, и в более раннем возрасте.
Миодистрофия Эрба-Рота (конечностно-поясная миодистрофия). Передается по аутосомно-рецессивному типу, оба пола страдают одинаково. Начало заболевания в большинстве случаев относится к середине 2-го десятилетия жизни (14-16 лет), однако описана как ранняя, псевдодюшенновская форма, когда первые симптомы проявляются в возрасте до 10 лет и заболевание протекает тяжело, так и поздний вариант с началом после 30 лет.
Течение заболевания может быть быстрым или более медленным, в среднем полная инвалидизация наступает через 15-20 лет от начала появления первых симптомов. Миодистрофия начинается либо с поражения мышц тазового пояса и проксимальных отделов ног (форма Лейдена-Мебиуса), либо с плечевого пояса (форма Эрба). В некоторых случаях плечевой и тазовый пояса поражаются одновременно.
Лечение мышечных дистрофий. Терапевтические возможности при мышечных дистрофиях весьма ограничены. Этиологического и патогенетического лечения практически не существует. Симптоматическое лечение направлено прежде всего на предотвращение развития контрактур, поддержание имеющейся мышечной силы и, возможно, на некоторое снижение скорости развития атрофии. Основная задача состоит в том, чтобы максимально продлить период, в течение которого больной способен самостоятельно передвигаться, так как в лежачем положении быстро нарастают контрактуры, сколиоз, дыхательные расстройства. Лечебный комплекс должен включать в себя лечебную гимнастику, массаж, ортопедические мероприятия, медикаментозную терапию.
Лечебная гимнастика состоит из пассивных и активных движений, выполняемых во всех суставах в различных положениях: стоя, сидя, лежа, при различном положении конечностей. Активные движения предпочтительнее выполнять в изометрическом режиме. Занятия гимнастикой необходимо проводить регулярно по нескольку раз в день. В то же время следует предостеречь от чрезмерных упражнений, особенно сопровождающихся перерастяжением мышц. Важное значение (особенно после иммобилизации больного) имеют дыхательные упражнения.
Ортопедические мероприятия консервативного (специальные шины) и оперативного характера (ахиллотомия, пересечение икроножной мышцы), направленные на коррекцию контрактур и формирующихся патологических установок конечностей, также имеют целью сохранить возможность самостоятельного передвижения. При этом в каждом случае необходимо индивидуально взвесить предполагаемую пользу и возможный вред от оперативного вмешательства. Следует учитывать, что нередко (в частности, при выраженном гиперлордозе и слабости четырехглавой мышцы бедра) эквиноварусная установка стоп имеет компенсаторное значение и после проведения, например, ахиллотомии больной может оказаться окончательно обездвиженным. При развивающихся контрактурах рекомендуется проводить осторожное растяжение мышц до 20-30 раз в день с последующим наложением шины на время сна.
Медикаментозная терапия предполагает назначение препаратов метаболического действия, направленных на восполнение энергетического и белкового дефицита, однако их эффективность весьма сомнительна. Применяют антагонисты кальция (в связи с выявленным при болезни Дюшенна дефектом клеточных мембран, приводящим к повышенному поступлению кальция внутрь клетки), иммуномодуляторы, фосфорсодержащие соединения (АТФ, фосфаден), витамин Е (100мг внутрь 3 раза в день). Показано, что при болезни Дюшенна применение преднизолона (0,75 мг/кг в сутки) может драматически увеличивать силу мышц, однако этот эффект сохраняется неболее года и в целом не влияет на исход заболевания. В связи с серьезными побочными эффектами, возникающими ври длительном применении препарата, его использование нецелесообразно. Оценки эффекта анаболических стероидов противоречивы и их назначение зачастую сопряжено с неоправданным риском. Оценивая эффект тех или иных препаратов при болезни Дюшенна, следует учитывать, что при умеренной выраженности заболевания у больных в возрасте 3-6 лет может отмечаться относительная стабилизация состояния, связанная с возрастным развитием мышечной системы, приобретением двигательных навыков, что может в какой-то степени временно скомпенсировать непрерывно текущий дистрофический процесс.
Определенное значение имеет коррекция питания больного, рекомендуется диета с высоким содержанием белка и низким содержанием жиров и пониженной калорийностью при оптимальном содержании витаминов и микроэлементов. Важную роль играет психологическая поддержка больного, продолжение обучения, правильная профессиональная ориентация.
biofile.ru
Мышечная дистрофия | Симптомы и лечение мышечной дистрофии
Симптомы мышечной дистрофии
Симптомы мышечной дистрофии имеют комплекс основных, базовых, проявлений, но и в зависимости от локализации и характеристик патологии имеются и собственные отличительные особенности. •
- В связи с дефицитом мышечной массы ног, наблюдаются нарушения в походке человека.
- Уменьшается тонус мышц.
- Атрофируются скелетные мышцы.
- Утрачиваются двигательные способности, которые были приобретены больным до момента начала прогрессирования заболевания: пациент перестает держать голову, ходить, сидеть, теряет и другие навыки.
- Притупляются болевые ощущения мышц, при этом чувствительность не пропадает.
- Снижение общего жизненного тонуса, больной начинает очень быстро утомляться.
- Мышечные волокна начинают замещаться соединительной тканью, что приводит к увеличению объемов самих мышц. Особенно это заметно по икроножному отделу.
- Проявляются трудности к обучению.
- Достаточно часты падения.
- Возникают сложности при беге и прыжках.
- Больному становиться тяжело вставать, как из лежачего положения, так и сидячего.
- Походка такого больного становится переваливающейся.
- Идет снижение интеллекта.
Мышечная дистрофия Дюшена
В настоящее время мышечная дистрофия дюшена является чаще всего проявляющейся разновидностью этого заболевания. Дефект, «благодаря» которому и развивается слабость мышечной ткани этого вида, найден и представляет собой модифицированный геном половой Х хромосомы. Зачастую, женщина, не болея сама, передает данный дефект своим детям. Первые симптомы патологии у мальчиков (ней, почему-то, преимущественно страдают именно они), получивших такой ген, выявляются уже в возрасте двух – пяти лет.
Первые признаки заболевания начинают проявляться в ослаблении тонуса нижних конечностей, а так же области таза. При дальнейшем прогрессировании заболевания подключаются к атрофии группы мышц верхней части тела. Постепенно, за счет перерождения мышечных волокон в соединительные, увеличиваются в объемах икроножные области нижних конечностей больного, увеличиваются размеры и жировой ткани. Темпы развития данного генетического нарушения достаточно велики и уже к 12 годам ребенок утрачивает способность вообще двигаться. Зачастую до двадцати лет такие пациенты не доживают.
Ослабление тонуса мышц нижних конечностей с разрастанием объемов икроножной области и приводит к тому, что ребенок начинает первично испытывать дискомфорт при ходьбе и беге, а впоследствии теряет эту способность полностью. Постепенно поднимаясь вверх и захватывая все большее количество групп мышц, на терминальной стадии мышечной дистрофии дюшена, патология начинает поражать и комплекс дыхательных мышц, глотки и лица.
Псевдогипертрофия может прогрессировать не только в икроножной области, она способна захватить и зоны ягодиц, живота и языка. При такой патологии достаточно часто возникает поражение и сердечных мышц (изменения идут по типу кардиомиопатии). Нарушается сердечный ритм, тона становятся глухими, само сердце увеличивается в размерах. Сердечная мышечная дистрофия, зачастую, и является причиной смерти пациента.
Характерной симптоматикой является и то, что больной страдает и умственной отсталостью. Это объясняется поражениями, которые захватывают и большие полушария головного мозга. При прогрессировании мышечной дистрофи, начинают появляться и другие сопутствующие заболевания. Такие, например, как: диффузный остеопороз, заболевания, связанные с эндокринной недостаточностью, идет деформация грудной клетки, позвоночника…
Основным отличительным признаком патологии по типу Дюшена от остальных видов, является большой уровень гиперферментемии, проявляющийся уже на начальной фазе развития патологии.
Прогрессирующая мышечная дистрофия
Наиболее часто, в области мышечно-неврологических заболеваний, встречается первичная прогрессирующая мышечная дистрофия, которая представлена достаточно обширной классификацией. Отличие одной формы от другой идет в зависимости от места генной мутации, скорости прогрессирования, возрастной характеристикой пациента (в каком возрасте начала проявляться патология), присутствует ли в симптоматике псевдогипертрофия и другие признаки. Большая часть из этих миодистрофий (их симптоматика), за практически вековую историю, достаточно неплохо изучена, но до сих пор патогенез так и не известен, а, исходя из этого, возникают проблемы и с высокой достоверностью диагностики. Не зная причин возникновения патологических изменений, очень тяжело и провести достаточно рациональную классификацию прогрессирующей мышечной дистрофии.
В большинстве своем, деление проводится либо по форме наследования, либо по клиническим характеристикам.
Первичная форма - повреждение мышечной ткани, при которой остаются действующими периферийные нервы. Вторичная форма – когда поражение начинается из нервных окончаний, изначально не затрагивая мышечные слои материю.
- Тяжелый тип псевдогипертрофии Дюшена.
- Реже встречающийся, менее агрессивный тип Беккера.
- Тип Ландузи - Дежерина. Затрагивает область плечо-лопатка-лицо.
- Тип Эрба – Рота. Подростковая форма заболевания.
Это основные типы мышечной дистрофии, которые диагностируются наиболее часто. Остальные разновидности встречаются реже и являются атипичными. Например, такие как:
- Дистрофия Ландузи Дежерина.
- Дистрофия Эмери Дрейфуса.
- Конечностно - поясная мышечная дистрофия.
- Окулофарингеальная мышечная дистрофия.
- А так же, некоторые другие.
Мышечная дистрофия Беккера
Эта патология встречается относительно редко и, в отличие от тяжелой злокачественной формы Дюшена, является доброкачественной и прогрессирует достаточно медленно. Одним из характерных признаков может служить то, что данной формой, как правило, болеют люди, имеющие небольшой рост. Достаточно длительное время болезнь не дает о себе знать и человек живет обычной жизнью. Толчком к развитию заболевания может явиться либо банальная бытовая травма, либо сопутствующее заболевание.
Мышечная дистрофия беккера относится к более легким формам данного заболевания и по остроте клинической симптоматики, и по полноте молекулярных проявлений. Симптомы в случае диагностики мышечной дистрофии по форме Беккера выявляются слабо. Больной с такой патологией способен достаточно нормально жить не один десяток лет. При такой слабой симптоматике дистрофию по Беккеру низко квалифицированный врач вполне может спутать с конечностно-поясничной дистрофией. Первые признаки данной патологии обычно начинают проявляться в двенадцатилетнем возрасте. Подросток начинает ощущать боли в нижних конечностях (в области голени), особенно во время нагрузки. Анализ мочи показывает высокое содержание миоглобина, являющегося показателем того, что в организме проходит распад мышечного белка. Идет повышение креатинкиназа в организме больного (фермента, вырабатывающегося из АТФ и креатина). Он активно используется организмом при возрастании физических нагрузок на него.
Симптоматика мышечная дистрофия беккера достаточно сильно перекликается с признаками, которые характеризуют и патологию по Дюшену. Однако проявления данной формы заболевания начинаются значительно в более поздний период (годам к 10 – 15), при этом прогрессирование болезни не настолько агрессивное. К тридцати годам такой пациент может еще не потерять трудоспособность и достаточно нормально ходить. Часты случаи, когда данная патология «идет по роду»: дед, болеющий данной болезнью, передает через свою дочку мутированный геном своему внуку.
Эта форма мышечной дистрофии была описана врачами и учеными Беккером и Кинером еще в 1955 году, поэтому и носит их имя (ее знают как мышечную дистрофию Беккера или Беккера-Кинера).
Симптоматика выявления патологии, как и в случае болезни по форме Дюшена, начинается с отклонений в тазово-поясной области, захватывая и нижние конечности. Это проявляется в изменении походки, появляются проблемы с подъемом по лестнице, очень тяжело такому больному становиться вставать из сидячего, на низких поверхностях, положения. Постепенно увеличиваются размеры икроножных мышц. При этом изменения области ахилловых сухожилий, заметные при патологии Дюшена, в данном случае визуализируются незначительно. Не наблюдается и снижения интеллектуальных способностей человека, что неизбежно при злокачественной мышечной дистрофии (по Дюшену). Не столь существенны и изменения в мышечной ткани сердца, поэтому при рассматриваемой болезни практически не наблюдается кардиомиопатия, либо она встречается в легкой форме.
Как и при других формах мышечной дистрофии, клинический анализ крови показывает повышения уровня некоторых ферментов в сыворотке крови, хотя они и не такие значительные как в случае с изменениями по Дюшену. Происходят сбои и в обменных процессах
Мышечная дистрофия Эрба Рота
Данную патологию называют еще юношеской. Симптомы этого заболевания начинают появляться в период с десяти до двадцати лет. Существенным отличием симптоматики данной формы заболевания является то, что первичным местом локализации изменений является плечевой пояс, а уже далее атрофия мышц начинает захватывать все новые области организма больного: верхние конечности, затем область пояса, таза и ног.
Случаи заболевания встречаются в пропорции 15 больных на один миллион населения. Дефектный геном переходит наследственно, по аутосомно-рецессивному пути. Страдают от этого заболевания, с равной вероятностью, как женщины, так и мужчины.
Мышечная дистрофия эрба рота существенно деформирует грудную клетку больного (как бы проваливая ее назад), живот начинает выдаваться вперед, походка становится неуверенной, переваливающейся. Первые признаки заболевания появляются приблизительно в 14 – 16 лет, но сам диапазон гораздо шире: бывают случаи и более позднего развития - после третьего десятка, или наоборот – лет в десять (при ранней симптоматике болезнь протекает с более тяжелыми проявлениями). Интенсивность и развитие течения болезни от случая к случаю различно. Но средняя продолжительность цикла с момента появления первых симптомов до полной инвалидности составляет от 15 до 20 лет.
Чаще всего мышечная дистрофия эрба начинает проявляться с изменений в тазово-поясном отделе, а так же с отеков и слабости в ногах. Далее распространяющаяся патология постепенно захватывает и остальные мышечные группы организма больного. Преимущественно поражение не затрагивает мышцы лица, сердечная мышца остается не тронутой, уровень интеллекта, обычно, держится на прежнем уровне. Количественный показатель ферментов в сыворотке крови слегка увеличен, но не до такого уровня как в предыдущих случаях.
Мышечная дистрофия рассматриваемой формы является одной из самых аморфных патологий.
Первичная мышечная дистрофия
Рассматриваемое заболевание является наследственным и связанным с полом (дефект генома Х-хромосомы). Путь передачи – рецессивный.
Клиника проявления достаточно ранняя – до трехлетнего возраста малыша. Даже в грудничковый период можно заметить отставание в развитии моторики у карапуза, позже, чем здоровые детки, они начинают сидеть и ходить. Уже к трем годам у малыша, заметна слабость в мышцах, он быстро устает, плохо переносит даже незначительные нагрузки. Постепенно атрофия захватывает тазовый пояс и проксимальные мышцы нижних конечностей.
Классикой симптоматики является псевдогипертрофия (мышечная материя заменяется жировой, увеличивая размеры данной области). Чаще такому поражению подвергается икроножная область, но встречаются случаи дефектности и дельтовидных мышц. Так называемые «икры гнома». Со временем малышу становится тяжело бегать и прыгать, подниматься вверх по лестнице. Через некоторое время, атрофия настегает и плечевой пояс.
Нервно-мышечная дистрофия
Медицина насчитывает ряд наследственных (генетических) заболеваний, поражающих мышечные и нервные ткани. Одна из них - нервно мышечная дистрофия, которая характеризуется нарушением моторных и статических проявлений на фоне мышечной атрофии. Поражению подвергаются нейроны, отвечающие за двигательные функции (клетки переднего рога), что приводит к изменениям группы тканей спинного мозга. Повреждение нейронов ядра клеток черепного нерва влияют на мимику лица, бульбарную и глазную мускулатуру. Так же за двигательные процессы отвечают однотипные клетки, при поражении которых страдают нервные окончания периферии и, нервно-мышечные соединения.
Базовые признаки такой патологии:
- Атрофия мышечно-соединительных тканей.
- Мышечные боли.
- Быстрая утомляемость больного.
- Снижение чувствительности рецепторов.
- Или наоборот повышенная чувствительность, вплоть до болевых синдромов.
- Появление внезапных судорог.
- Головокружения.
- Патология сердца.
- Ухудшение зрения.
- Сбой в системе потоотделения.
Мышечная дистрофия Ландузи Дежерина
Чаще всего патология данной формы начинает проявляться у подростков в 10 – 15 лет, хотя фактически известны случаи, когда мышечная дистрофия ландузи дежерина начинала развиваться у шестилетних детей, либо у пятидесятилетнего человека. Первичной областью патологии, чаще всего, является группа мышц лицевой зоны. Постепенно ореол поражения расширяется, начинают атрофироваться группы плечевого пояса, торса и далее вниз. При поражении лицевой мимики в ранний период заболевания, неплотно закрываются веки. Приоткрытыми остаются и губы, что приводит к речевому дефекту. Течение болезни идет медленно – на протяжении этого периода человек абсолютно трудоспособен, только лет через 15 – 20 постепенно начинают атрофироваться мышцы пояса и таза – это и приводит уже к двигательной пассивности. И только к 40 – 60 годам поражение полностью захватывает нижние конечности.
То есть мышечную дистрофию ландузи дежерина можно назвать благоприятно текущим проявлением мышечного поражения.
Мышечная дистрофия Эмери Дрейфуса
Как и все предыдущие, мышечная дистрофия эмери дрейфуса является заболеванием наследственным. Основная зона поражения – атрофия плечелоктевых и голеностопных мышц. Данная болезнь характеризуется длительным периодом развития. В подавляющем большинстве случаев поражению подвергается сердце: брадиаритмияи, снижение кровяной проходимости, блокада и другие. Сбои в работе сердца могут быть причиной обмороков, а иногда даже летального исхода.
Ранняя диагностика не только самого заболевания, а и дифференцирование ее формы, поможет спасти жизнь не одному больному.
Конечностно-поясная мышечная дистрофия
Конечностно поясная мышечная дистрофия относится к наследственной патологии, путями наследования которой являются как аутосомно-рецессивные, так и аутосомно-доминантные болезни. Базовый район поражения – это область пояса, торса и верхних конечностей. При этом мышцы лицевой мускулатуры не страдают.
По данным исследований удалось установить как минимум два локуса генома хромосом, при мутации которых создается толчок к развитию конечностно поясной мышечной дистрофии. Прогрессирование данного поражения проходит достаточно медленно, давая больному в полной мере насладиться жизнью.
Окулофарингеальная мышечная дистрофия
Аутосомно-доминантная болезнь, проявляющаяся уже в достаточно зрелом возрасте - окулофарингеальная мышечная дистрофия. Как не странно это звучит, но данная патология поражает людей, принадлежащих определенным этническим группам.
Чаще всего симптоматика начинает проявляться к 25 – 30 годам. Классическими признаками данной мышечной дистрофии является атрофия лицевых мышц: птоз век, проблемы с глотательной функцией (дисфагия). Болезнь, постепенно прогрессируя, приводит к неподвижности глазного яблока, при этом внутренние мышцы глаза не подвергаются поражению. На этом этапе изменения могут остановиться, но иногда патологии подвергаются и остальные лицевые мышцы. Достаточно редко, но бывают задействованы в разрушительном процессе и группы мышц плечевого пояса, шеи, неба и глотки. В этом случае кроме офтальмоплегии и дисфагии прогрессирует еще и дисфония (проблема речевого аппарата).
Мышечная дистрофия у детей
Детство. Многие его вспоминают с улыбкой. Прятки, качели, велосипеды… Да сколько еще различных игр придумывает детвора. Но есть малыши, которые не могут себе позволить такой роскоши. Мышечная дистрофия у детей не дает такой возможности.
Практически все, за редким исключением, формы могут проявляться у деток своей симптоматикой: и злокачественная форма патологии по Дюшону (развивающаяся только у мальчиков), и доброкачественная мышечная дистрофия по Беккеру и другие. Особенно опасна патология, развивающаяся стремительно, агрессивно (форма по Дюшону). Причем для малыша опасна даже не столько сама симптоматика (атрофия практически всех групп мышц), как вторичные осложнения, которые и приводят к двадцати годам к смерти. Чаще всего летальный исход наступает вследствие респираторной инфекции или сердечной недостаточности. Но данная симптоматика становится более явной только тогда, когда малыш начинает делать первые шаги.
- Задержка в развитии: такие дети позже начинают сидеть и ходить.
- Медленное интеллектуальное развитие.
- Первыми поражаются мышцы позвоночника.
- Таким малышам трудно бегать и подниматься по лестнице.
- Походка вперевалочку.
- Деформация позвоночника.
- Ходьба на пальцах.
- Малышу тяжело держать свой вес, и он быстро утомляется.
- За счет жировой ткани увеличивается размер мышц.
- Поражение конечностей идет симметрично.
- Патологическое увеличение челюсти и промежутков между зубками.
- Приблизительно с 13 лет малыш перестает ходить совсем.
- Патология сердечной мышцы.
При других формах поражения, симптоматика достаточно похожа, только тяжесть поражения значительно ниже.
ilive.com.ua
Прогрессирующие мышечные дистрофии - Болезни неврологии
ссылки Наиболее распространенными среди нервно-мышечных заболеваний являются первичные мышечные дистрофии. Различные формы миодистрофий отличаются друг от друга типами наследования, сроками начала процесса, характером и быстротой его течения, своеобразием топографии мышечных страданий, наличием или отсутствием псевдогипертрофий и сухожильных ретракций и другими признаками.Большинство мышечных дистрофий достаточно хорошо изучено клинически, их подробное описание сделано еще в конце прошлого века. Но, несмотря на почти вековую историю изучения миодистрофий, вопросы их патогенеза, достоверной диагностики и лечения остаются до сего времени неразрешенными. Существует большое количество классификаций, но отсутствие точных данных о первичном биохимическом дефекте не дает возможности построить ее по рациональному принципу. В имеющихся классификациях основой являются или клинический принцип, или тип наследования. Так, Walton (1974) предлагает различать следующие формы миодистрофий.A. Х-сцепленные мышечные дистрофии:а) тяжелая (тип Дюшенна)б) благоприятная (тип Беккера)B. Аутосомно-рецессивные мышечные дистрофии:а) конечностно-поясная или ювенильная (тип Эрба)б) детская мышечная дистрофия (псевдодюшенновская)в) врожденные мышечные дистрофииC. Лицелопаточно-плечевую (Ландузи - Дежерина)D. Дистальную мышечную дистрофиюE. Окулярную мышечную дистрофиюF. Окулофарингеальную мышечную дистрофиюПоследние несколько форм относятся к аутосомно-доминантным типам наследственной передачи с высокой или неполной пенетрантностью. Следует подчеркнуть, что диагностика мышечных дистрофий нередко представляет большие трудности. Имеется большая вариабельность клинических проявлений, а малое число детей в семье затрудняет определение типа наследования. Наиболее часто встречаются миодистрофии Дюшенна, Эрба и Ландузи - Дежерина.В настоящее время выявлена значительная группа непрогрессирующих миопатий, которые являются своеобразным пороком развития на уровне мышечной клетки.Мышечная дистрофия типа Дюшенна
Псевдогипертрофическая мышечная дистрофия Дюшенна - наиболее хорошо изученная форма, встречается чаще других заболеваний мышечной системы (3,3:100 000 населения). Она характеризуется ранним началом и злокачественным течением. Классическая картина проявляется изменением походки у ребенка в возрасте 2-5 лет, к 8-10 годам дети ходят уже с трудом, к 14-15 годам они, как правило, полностью обездвижены. У некоторых детей начальные симптомы проявляются отставанием двигательного развития: они позднее начинают ходить, не умеют бегать и прыгать, при ходьбе отмечается некоторое раскачивание.Одними из первых признаков заболевания являются уплотнение икроножных мышц и постепенное увеличение их объема за счет псевдогипертрофий. Локальные атрофии мышц бедер, тазового пояса нередко маскируются хорошо развитым подкожным жировым слоем. Постепенно процесс принимает восходящее направление и распространяется на плечевой пояс, мышцы спины, а затем и на проксимальные отделы рук. В терминальных стадиях слабость мышц может распространяться на мышцы лица, глотки, дыхательные мышцы.В развитой стадии болезни имеются такие характерные симптомы, как «утиная походка», подчеркнутый поясничный лордоз, «крыловидные лопатки», симптом «свободных надплечий». Довольно типичны ранние мышечные контрактуры и сухожильные ретракции, особенно ахилловых сухожилий. Рано выпадают коленные рефлексы, а затем рефлексы с верхних конечностей.Псевдогипертрофии могут развиваться не только в икроножных, но также в ягодичных, дельтовидных мышцах, мышцах живота, языка. Очень часто страдает сердечная мышца по типу кардиомиопатии с изменением ЭКГ на ранних стадиях патологического процесса. При обследовании выявляются нарушение ритма сердечной деятельности, расширение границ сердца, глухость тонов. Острая сердечная слабость - наиболее частая причина летальных исходов при миодистрофии Дюшенна. На вскрытии находят фиброз и жировую инфильтрацию сердечной мышцы.Довольно характерным симптомом заболевания является снижение интеллекта. Дюшенн, впервые описавший эту форму, обратил внимание на умственную отсталость больных детей. Представляет интерес тот факт, что в одних семьях олигофрения бывает резко выражена, в других - сравнительно умеренно. Изменение высших психических функций не может быть объяснено только педагогической запущенностью больных детей (они рано выключаются из детских коллективов, не посещают детский сад и школу из-за двигательного дефекта). Патоморфологическое исследование после летального исхода выявляет изменения в строении извилин больших полушарий, нарушение цитоархитектоники мозговой коры; ПЭГ у больных показывает развитие гидроцефалии. Нередко у детей развивается адипозогенитальный синдром, иногда и другие признаки эндокринной недостаточности. Часто находят изменения в костной системе: деформацию стоп, грудной клетки, позвоночника, диффузный остеопороз. Отличительной особенностью формы Дюшенна, выделяющей ее из остальных мышечных дистрофий, является высокая степень гиперферментемии уже на ранних стадиях развития процесса. Так, уровень специфического для мышечной ткани фермента - креатинфосфокиназы - в сыворотке крови может превышать в десятки и даже сотни раз нормальные показатели. Также значительно повышается активность альдолаз, лактатдегидрогеназы и других ферментов. Лишь в далеко зашедших стадиях болезни степень гиперферментемии постепенно снижается. Имеются сообщения о повышении креатинфосфокиназы на стадии внутриутробного развития. При болезни Дюшенна изменяется обмен креатина. Уже на сравнительно ранних этапах заболевания обнаруживается креатинурия и резко уменьшается экскреция с мочой креатинина. Последний показатель более постоянен и степень уменьшения выделения креатинина в известной степени говорит о тяжести и остроте дистрофического процесса. Наблюдается также увеличение экскреции с мочой аминокислот.Миодистрофия Дюшенна передается по рецессивному, сцепленному с Х-хромосомой типу. Довольно высока частота мутации гена, что объясняет большое количество спорадических случаев. Для медико-генетического консультирования очень важно установление гетерозиготного носительства. При миодистрофии Дюшенна у заведомых гетерозиготных носителей приблизительно в 70% случаев выявляются субклинические, а иногда и явные признаки мышечной патологии - некоторое уплотнение и даже увеличение икроножных мышц, быстрая утомляемость мышц при интенсивной Физической нагрузке, небольшие изменения на ЭМГ и при патоморфологическом изучении мышечных биоптатов. Наиболее часто у гетерозиготных носительниц выявляется увеличение активности ферментов в сыворотке крови, в частности повышение активности креатинфосфокиназы. Наличие клинических или субклинических признаков заболевания можно объяснить гипотезой Мэри Лайон [Lyon, 1962], согласно которой сумма клеток, содержащих неактивную Х-хромосому с нормальным геном, больше, чем содержащих мутантный ген.При наличии клинической картины миодистрофии Дюшенна у лиц женского пола следует в первую очередь исключить возможность аномалии по Х-хромосоме - синдром Шерешевского - Тернера (ХО), синдром Морриса (XY) или мозаицизм по этим синдромам.Мышечная дистрофия типа Беккера-Кинера
Наряду с тяжелой, злокачественной формой Х-сцепленной миодистрофии (тип Дюшенна) существует доброкачественная форма заболевания (тип Беккера - Кинера). По клиническим симптомам она очень напоминает форму Дюшенна, однако начинается, как правило, позднее - в 10-15 лет, течет мягког больные длительно сохраняют работоспособность, в возрасте 20-30 лет и позже еще могут ходить, фертильность не снижена. Заболевание прослеживается в нескольких поколениях семьи, нередко имеется так называемый «эффект деда» - больной мужчина через свою дочь передает заболевание внуку. Впервые доброкачественная форма Х-сцепленной миодистрофии была описана в 1955 г. Becker и Kiner. Начальные симптомы, как и при болезни Дюшенна, проявляются слабостью в мышцах тазового пояса, затем в проксимальных отделах нижних конечностей. У больных изменяется походка, они испытывают затруднение при подъеме по лестнице, при вставании с низкого сиденья. Характерны псевдогипертрофии икроножных мышц. Ретракции ахилловых сухожилий выражены менее резко, чем при болезни Дюшенна. При этой форме не отмечается нарушений интеллекта, почти не встречается кардиомиопатия или она выражена незначительно. Как и при других Х-сцепленных миодистрофиях, при форме Беккера - Кинера изменяется уровень ферментов в сыворотке крови - значительно повышается активность креатинфосфокиназы, лактатдегидрогеназы и альдолазы, хотя и в меньшей степени, чем при болезни Дюшенна. Нарушается также обмен креатина и аминокислот. В литературе обсуждается вопрос о нозологической самостоятельности болезни Беккера - Кинера. Не решен окончательно вопрос о том, определяются ли формы Беккера - Кинера и Дюшенна различными мутантными аллелями в том же самом генном локусе или в двух различных локусах. McKusick (1962) предполагает, что имеется несколько форм Х-сцепленных мышечных дистрофий, так же как имеется несколько форм цветовой слепоты, гемофилии и дегенерации сетчатки.Существенным доказательством в пользу нозологической самостоятельности доброкачественной формы заболевания являются некоторые биохимические исследования. Так, было показано, что при миодистрофии Дюшенна происходит высокий коллагеновый и низкий неколлагеновый синтез белка в тяжелых полирибосомах, а при миодистрофии Беккера - Кинера увеличен как коллагеновый, так и неколлагеновый синтез в полисомах. Патоморфологические исследования также выявляют известные различия - при форме Беккера-Кинера имеется отчетливая сохранность регенераторных процессов в мышечной ткани, кроме того, сохранена миоглобинпероксидазная активность в противоположность болезни Дюшенна, где последняя постоянно резко снижена.При изучении групп сцепления в Х-хромосоме было показано, что локус глюкозо-6-фосфатдегидрогеназы и локус доброкачественной формы Беккера - Кинера находятся ближе, чем локус злокачественной формы Дюшенна. Однако исследования были сделаны только на трех семьях с доброкачественной формой.Против нозологической самостоятельности этих двух заболеваний говорят накапливающиеся описания семей, в которых имеется сочетание больных с обеими формами. Так,. Walton (1956) описал семью, где 3 брата с болезнью Дюшенна имели 3 дядей по материнской линии, страдающих доброкачественной формой миодистрофии. Furukawa с соавт. (1977) наблюдал 3 семьи, в которых сосуществовали обе формы. Учитывая различное течение и прогноз этих двух форм, рациональнее их оценить как разные заболевания.Редкие формы Х-сцепленных миодистрофий
В настоящее время известно несколько вариантов сравнительно редко встречающихся наследственных мышечных дистрофий, передающихся через Х-хромосому и (как при форме Беккера-Кинера) имеющих мягкое, благоприятное течение. К таким формам относятся: миодистрофия Дрейфуса - Хогана, форма Мэбри, форма Роттауфа - Мортье - Бейера, формы Робера и Хэка-Лаудана. Форма Дрейфуса-Хогана описана в 1961 г. По срокам начала она напоминает болезнь Дюшенна, чаще это 4-5-летний возраст. Развиваются мышечная слабость и атрофии в мышцах тазового пояса и проксимальных отделов нижних конечностей. Очень медленно процесс распространяется на мышцы плечевого пояса и проксимальных отделов верхних конечностей, иногда вовлекается мускулатура лица, в частности круговая мышца рта. Характерной особенностью этой формы является отсутствие псевдогипертрофий и раннее развитие сухожильных ретракций в ахилловых сухожилиях, л также в сухожилиях двуглавой мышцы плеча и других. Интеллект больного сохранен. Нередко развивается кардиомиопатия с изменением сердечного ритма, чаще всего в возрасте 30-40 лет. Цветовое зрение нормально. Значительно повышается активность ферментов сыворотки крови, особенно возрастает уровень креатинфосфокиназы; в далеко защедших стадиях ферментемия постепенно снижается.Форма Мэбри известна с 1965 г. Автор и сотрудники наблюдали семью, где в 2 поколениях у 9 лиц мужского пола имелась характерная клиническая картина. Первые симптомы появлялись в пубертатном периоде (11-13 лет) в виде слабости в мышцах бедер и тазового пояса. Имелись выраженные псевдогипертрофии. Для этой формы миодистрофии не характерны сухожильные ретракции, нет цветовой спепоты и других маркеров по Х-хромосомной патологии. Интеллект сохранен. Постоянно страдает мышца сердца. Повышена активность сывороточных ферментов.При биопсии мышцы выявляются выраженные атрофические изменения с уменьшением размеров мышечных волокон и отсутствием гипертрофированных. Уменьшено количество коллагеновых волокон и резко выражен липоматоз.Форма Роттауфа - Мортье - Бейера описана впервые в 1971 г. Авторы наблюдали большую семью, где в 4 поколениях было 17 больных мужчин. Характерной особенностью этой формы является развитие ранних и резко выраженных сухожильных ретракций и мышечных контрактур. Эти симптомы появляются в возрасте 5-10 лет вначале в дистальных отделах ног (ограничение тыльного сгибания стоп), затем развивается ограничение сгибания шеи и разгибания в локтевых суставах. Постепенно формируются патологические позы головы и туловища из-за прогрессирующего фиброза мышц с невозможностью сгибания позвоночника. Парезы выражены очень умеренно, в основном в мышцах плечевого пояса, а также в дистальных отделах ног; мышечные гипотрофии диффузные, но нерезкие. Псевдогипертрофии полностью отсутствуют.Интеллект у больных сохранен (среди них встречаются даже одаренные люди). Грубо страдает сердечная мышца, как правило, постепенно формируется нарушение проводимости и к 35-40 годам может развиться полная атриовентрикулярная блокада. ЭМГ и данные биопсии указывают на миогенный характер изменений. Отмечается четкая гиперферментемия, степень которой снижается в далеко зашедших стадиях процесса. У гетерозиготных носительниц нет клинических проявлений и показатели активности ферментов нормальны.Прогрессирование заболевания очень медленное, больные длительно сохраняют возможность самообслуживания и даже трудоспособность. Многие вступают в брак и могут иметь детей. Фертильность не ограничена. Летальный исход, как правило, наступает в возрасте 40-50 лет и обусловлен поражением сердечной мышцы.Конечностно-поясная форма мышечной дистрофии (ювенильная миопатия Эрба)
Встречается с частотой 1,5:100 000 населения. Передается по аутосомно-рецессивному типу наследования, оба пола страдают одинаково часто.Начало заболевания в большинстве случаев относится к середине второго десятилетия жизни (14-16 лет), однако имеется довольно широкий возрастной диапазон. Описана так называемая ранняя, или псевдодюшеновская, форма, когда первые симптомы проявляются в возрасте до 10 лет и течение заболевания более тяжелое. Имеется и поздний вариант с началом после 30 лет.Течение заболевания может быть быстрым или более медленным, в среднем полная инвалидность наступает через 15-20 лет от начала появления первых симптомов. В большинстве случаев миодистрофия Эрба начинается с поражения мышц тазового пояса и проксимальных отделов ног, где появляются слабость и похудание мышц. Процесс в дальнейшем распространяется на плечевой пояс. В некоторых случаях плечевой п тазовый пояса поражаются одновременно. Довольно значительно страдают мышцы спины и живота. У больных имеется характерная «утиная» походка, затруднено вставание из положения лежа и сидя, подчеркнут поясничный лордоз. Мышцы лица в большинстве случаев не страдают. Для этой формы миодистрофии сравнительно мало характерны значительные контрактуры и псевдогипертрофии. Могут иметь место концевые атрофии и сухожильные ретракции. Интеллект у больных обычно сохранен. Сердечная мышца большей частью не поражена. Уровень ферментов в сыворотке крови, как правило, повышен, однако далеко не столь резко, как при Х-сцепленной миодистрофии. Есть указания, что у больных мужского пола уровень креатинфосфокиназы выше, чем у больных женщин. Имеется значительная разница в экспрессивности мутантного гена у разных членов семьи - наряду с тяжелой клинической картиной могут быть сравнительно легкие и даже стертые клинические симптомы. Нарушается креатин-креатининовый обмен, особенно резко уменьшается экскреция креатинина, увеличено выделение альфааминоазота с мочой. На ЭМГ находят изменения миогенного типа со снижением амплитуды биопотенциалов и сохранной частотой.Миодистрофия Эрба - наиболее аморфная форма и большинство фенокопий имитирует именно эту форму патологии, поэтому очень важно в спорадических случаях проводить тщательное клиническое обследование для исключения в первую очередь воспалительного поражения мышц типа полимиозита, особенно при наличии болевого синдрома, а также эндокринных миопатий, токсических, лекарственных, карциноматозных и других миопатий. Подобные фенокопии встречаются особенно часто в пожилом возрасте.Лицелопаточно-плечевая форма миодистрофии (тип Ландузи-Дежерина)
Эта форма миодистрофии описана в 1884 г. Landousi и Dejerine. Встречается реже, чем две предыдущие формы (0,9: 100ООО населения). Заболевание передается по регулярному аутосомно-доминантному типу с высокой пенетрантностью и несколько вариабельной экспрессивностью. По данным некоторых авторов, женщины болеют чаще мужчин (3: 1). Физические перегрузки, интенсивные занятия спортом, а также нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни.Миодистрофия Ландузи--Дежерина - сравнительно благоприятно текущая форма мышечной патологии. Начинается она чаще в возрасте около 20 лет, иногда позже. Однако в семейных случаях заболевания, когда можно проследить за младшими членами семьи в динамике удается выявить некоторую слабость мышц, например лица, в более раннем возрасте.По-видимому, вначале слабо выраженные симптомы длительное время остаются стабильными, а затем наступает нрогредиентность течения. Больные доживают до солидного иозраста (60 лет и более).Мышечная слабость и атрофии вначале появляются в мышцах лица или плечевого пояса. Постепенно эти нарушения распространяются на мышцы проксимальных отделов рук, а затем и на нижние конечности. Характерно, что в большинстве случаев вначале поражаются мышцы передней поверхности голеней, затем мышцы проксимальных отделов ног. На высоте заболевания грубо страдают круговые мышцы глаза н рта, большая грудная, передняя зубчатая и нижние отделы трапециевидной мышц, широкая мышца спины, двуглавая, трехглавая мышцы плеча. Характерен внешний вид таких -больных: типичное лицо «миопата» с «поперечной улыбкой», резко выраженные «крыловидные лопатки», своеобразная деформация грудной клетки за счет мышечного скелета с уплощением ее в переднезаднем направлении и ротацией внутрь плечевых суставов. Нередко имеется асимметрия поражения, даже в пределах одной мышцы (например круговой мышцы рта). Наблюдаются псевдогипертрофии икроножных, дельтовидных мышц, иногда мышц лица. Контрактуры и ретракции выражены умеренно. Сухожильные рефлексы длительное время бывают сохранены.Признаки поражения сердечной мышцы выявляются редко и они практически не отличаются от таковых в общей популяции, хотя нарушения атриовентрикулярной проводимости описаны. Уровень активности сывороточных ферментов увеличен незначительно, может быть даже нормальным. Креатин-креатининовый обмен нарушен умеренно, хотя некоторое уменьшение креатинина в моче выявляется постоянно. Интеллект у больных при этой форме не страдает. Представляет интерес тот факт, что ЭМГ у больных миодистрофией Ландузи - Дежерина нередко не совсем типична для мышечного уровня поражения. У некоторых больных (членов одной семьи) может наблюдаться характерное снижение амплитуды биопотенциалов, интерференционный тип кривой, у других, наоборот, уменьшение частоты и гиперсинхронная активность, иногда с типичным ритмом частокола. Следует помнить о наличии неврогенного варианта плечелопаточно-лицевой миодистрофии.В настоящее время ряд авторов полагают, что форма Ландузи-Дежерина не является единой, гомогенной формой, а представляет собой синдром. Лицелопаточно-плечевой синдром встречается при миодистрофии Ландузи - Дежерина, неврогенной амиотрофии, при миастении, миотубулярной, немалиновой миопатии, митохондриальной миопатии и центроядерной миопатии. Клинический диагноз должен подкрепляться, помимо электромиографического исследования, результатами гистохимического и электронно-микроскопического исследований.Дистальная форма мышечной дистрофии
Первое сообщение об этой форме мышечного поражения относится к 1907 г. Spiller привел клинические и патологоанатомические данные и отметил, что заболевание отличается от невральной амиотрофии Шарко - Мари. Подробное клиническое описание дистальной формы мышечной дистрофии дано в 1951 г. Welander, которая наблюдала боле 250 больных в Швеции. Заболевание встречается сравнительно редко. Тип наследования - аутосомно-доминантный с неполной пенетрантностью и вариабельной экспрессивностью.Первые симптомы заболевания проявляются в сравнительно позднем возрасте, как правило, позже 20 лет, хотя имеются описания болезни с началом в 5-15 лет [Pratt, 1967]. Заболевание отличается доброкачественным течением. Поражаются дистальные отделы нижних конечностей - появляются парезы стоп, голеней, развивается мышечное похудание. Постепенно слабость и гипотрофии распространяются на кисти и предплечья, в далеко зашедших случаях могут страдать проксимальные отделы ног. Выпадают сначала ахилловы рефлексы, затем коленные и рефлексы на руках. Не наблюдается псевдогипертрофий, фасцикуляций, всегда сохранена чувствительность. Сухожильные ретракции также мало характерны. В очень редких случаях развивается кардиомиопатия.Заболевание не всегда легко разграничить с невральной амиотрофией Шарко - Мари. Опорными пунктами в диагностике служат данные электрофизиологических методов исследования. При дистальной миопатии всегда нормальна скорость проведения возбуждения по нервному стволу, ЭМГ указывает на мышечный тип поражения. Следует отметить, что наблюдается неврогенная амиотрофия с локализацией парезов и похуданий мышц в дистальных отделах рук и ног. В этих случаях ЭМГ регистрирует типичный спинальный характер биоэлектрической активности с урежением частоты и явлениями синхронизации. Важным диагностическим критерием является исследование сывороточных ферментов, активность которых может значительно увеличиваться при мышечной дистрофии и не меняться при спинальной и невральной амиотрофии. Четкая креатинурия и резкое снижение экскреции с мочой креатинина будут свидетельствовать также в пользу миогенной природы страдания.Окулярная и окулофарингеальная миопатии
Изолированное первичное поражение мышц глазного яблока впервые отмечено Говерсом и Мебиусом около 100 лет тому назад, однако подробное описание этой формы поражения дано в 1951 г. Kilon. Заболевание встречается редко. Тип наследственной передачи аутосомно-доминантный, с низкой пенетрантностью. Часто имеют место спорадические случаи.Начало заболевания в возрасте 25-30 лет, но иногда первые симптомы отмечают в пубертатном периоде. Вначале появляются небольшой птоз, который постепенно увеличивается, затем ограничение движений глазного яблока, как правило, симметричное. Жалобы на двоение в глазах крайне редки. Течение заболевания медленно прогрессирующее, обычно до полной наружной офтальмоплегии. Внутренние мышцы глаза не страдают. Процесс на этом иногда приостанавливается, однако в ряде случаев присоединяется слабость круговой мышцы глаза, лобной мышцы и других мимических мышц. На ЭМГ и при исследовании биоптата обнаруживается заинтересованность мышц шеи и плечевого пояса; иногда парезы и гипотрофия этих мышц выявляются и клинически. В редких случаях отмечается широкая генерализация процесса.При окулофарингеальной миопатии, которая встречается еще реже, в процесс включаются также мышцы глотки и мягкого неба. Это заболевание встречается после 40 лет. В подобных случаях, помимо офтальмоплегии, развивается дисфагия и дисфония.При патоморфологическом исследовании выявляются разнокалиберность мышечных волокон, наличие маленьких ангулярных волокон, вакуолярные изменения. Разрастание соединительной ткани, фагоцитоз и базофилия встречаются нечасто. Во многих случаях находят измененные митохондрии, которые нередко увеличены в размере, кристы в них располагаются неправильно - по периферии.Дифференциальный диагноз в ряде случаев труден с особой формой глазной миастении. Этой формой миастении болеют чаще лица мужского пола, начало ее нередко острое, возраст больных от 20 до 30 лет. Характерно течение без ремиссий, имеет место резистентность к антихолинэстеразным препаратам. Решающим в диагностике является электромиографическое исследование с ритмической стимуляцией и проведением тестов с кураре или тензилоном.Проводят также дифференциальный диагноз с органическими поражениями головного мозга (опухоли среднего мозга, воспалительные процессы мозга и его оболочек).Редкие формы прогрессирующих мышечных дистрофий. Описано значительное число больных с врожденной мышечной дистрофией, у которых имелась картина «floppy baby» [Walton, 1954, 1967, 1968]. У части таких больных диффузная мышечная слабость, гипотония, обнаруживаемые при рождении, могут сочетаться с множественными контрактурами (тип артрогрипоза). Дети с подобными формами рано умирают. Тип наследования аутосомно-рецессивный.К редким формам мышечных дистрофий относится миопатия четырехглавых мышц бедра и ряд других миопатий.Патоморфологнческие изменения при прогрессирующих мышечных дистрофиях
Изменения со стороны нервной системы при мышечных дистрофиях отсутствуют или они минимальны. Описана патология спинного мозга, в котором иногда находят уменьшение клеток передних рогов. Отмечают изменения двигательных нервных окончаний (осевых цилиндров и миелиновых оболочек). Отмечено нарушение структуры моторных бляшек с исчезновением фибриллярной структуры [Jerusalem et al., 1974].Основные изменения отмечены в самой мышечной ткани. Мышечные волокна истончаются, замещаются жировой и соединительной тканью, отдельные волокна гипертрофируются, увеличивается количество мышечных ядер. Последние могут располагаться цепочками. Находят изменения в сосудах - утолщение стенок, стенозирование, иногда наблюдаются микротромбозы. Гистохимическое исследование мышечного биоптата позволяет выявить накопление кислых мукополисахаридов, уменьшение ряда ферментов. При электронно-микроскопическом изучении было установлено разрушение миофиламентов, расширение межфибриллярных пространств, изменение z-полос, увеличение каналов саркоплазматического ретикулума с образованием вакуолей. Изменяется структура митохондрий, они могут приобретать шаровидную форму, атрофируются кристы, как правило, увеличивается число лизосом.Патогенез прогрессирующих мышечных дистрофий
Вопросам изучения патогенеза прогрессирующих мышечных дистрофий посвящено огромное количество исследований, однако до сего времени первичный биохимический дефект не открыт, не выяснены механизмы гибели мышечных волокон, неизвестны причины избирательного поражения мышц при различных формах миодистрофии. На современном этапе не утратили своего значения следующие гипотезы: неврогенная, гипоксическая, дефектных мембран, дисфункции внутриклеточных медиаторов.Неврогенная гипотеза предполагает первичное поражение нервной системы (спинного мозга, а также периферических ее отделов, включая внутримышечные волокна) с вторичным нарушением метаболизма мышечной ткани. В основе этой гипотезы лежат данные, указывающие на уменьшение мотонейронов в передних рогах спинного мозга, наличие дегенеративных изменений в нервных терминалях и концевых пластинках, уменьшение количества моторных единиц в дистрофических мышцах, изменение аксонального тока структурных белков и низкомолекулярных соединений, некоторое замедление проводимости возбуждения в дистальных отделах нервных стволов. Эти данные являются новым подкреплением старых представлений о патогенезе мышечных дистрофий как следствие нарушения трофической функции нервной системы, в частности ее симпатического отдела.Особый интерес представляет концепция поломки бета-адренорецепторов, что может привести к снижению чувствительности к действию медиаторов вегетативной системы - адреналину и норадреналину и к автономии обмена в мышечной ткани [Хохлов А. П., 1977; Mawatary, 1975]. Несмотря на кажущуюся стройность неврогенной гипотезы, первичность выявленных изменений полностью не доказана. Так, компьютерный метод подсчета моторных единиц не установил существенных различий между нормальной и дистрофичной мышцами. Исследование передних рогов спинного мозга при вскрытии умерших от миодистрофии Дюшенна не выявило патологии. Изменение нервных терминалей, а также аксонального тока веществ может быть вторичным как результат грубого дистрофического процесса в мышце с восходящим перерождением нервных волокон. С позиций неврогенной гипотезы нельзя объяснить клинические и биохимические особенности разных форм миодистрофий. Однако все это не исключает участия нервной системы в общем комплексе патогенетических механизмов.Гипотеза тканевой гипоксии объясняет гибель мышечных волокон как следствие хронического недостатка кислорода. Предпосылками для этой гипотезы явились патоморфологические данные о сходстве изменений мышц у животных с экспериментальной гипоксией и у больных с миодистрофиями, создание экспериментальной модели миопатии с помощью искусственной эмболизации частицами декстрана, а также повторными инъекциями смеси имипрамина и серотонина. Увеличение кислых мукополисахарндов в основном веществе мышц и стенках сосудов с прогрессирующим новообразованием коллагеновых волокон, а затем формированием плотного фиброзного футляра вокруг мышечных волокон, последующим сдавленней сосудов, хроническим нарушением микроциркуляции (Ситников В. Ф., 1973, 1976] на ранних стадиях миодистрофического процесса служит известным доказательством данной гипотезы. Изучение обмена миоглобина, показавшее его дефектность (близость к фетальному, т. е. функционально неполноценному), еще больше подкрепило гипотезу тканевой гипоксии как первопричины развивающейся гибели мышечной ткани.Однако последующие контрольные исследования с применением более современных методов не смогли полностью подтвердить эту гипотезу. Так, измерение мышечного кровотока с помощью радиоактивного ксенона выявило нормальный уровень. Электронно-микроскопическое исследование не подтвердило наличия сосудистой окклюзии, а морфометрический анализ капилляров показал их нормальное количество. Повторные эксперименты с эмболизацией сосудов суспензией сухих частиц декстрана не дали возможности получить модель, характерную для миопатии.Не находит объяснения с позиций этой гипотезы механизм гибели белых мышечных волокон, хотя источником энергии в них является анаэробный гликогенолиз. Не было также получено доказательств разобщения тканевого дыхания и окислительного фосфорилирования.Гипотеза дефектных мембран. Согласно этой гипотезе, первичным в патогенезе мышечных дистрофий является увеличение проницаемости сарколеммы, а также субклеточных мембран - лизосомальных, митохондриальных, саркотубулярных, в связи с чем происходит потеря таких веществ, как внутриклеточные ферменты, гликоген, аминокислоты, креатин и т. д. Все это приводит к уменьшению количества жизненно важных белков, нарушению баланса биохимических процессов. Обнаружение подобных сдвигов у гетерозиготных носителей подтверждает первичный характер этих структурных нарушений. Однако ряд фактов не находит объяснения с позиций данной гипотезы. Так, было показано, что нарушение проницаемости мембран имеет избирательный характер - такие вещества, как миоглобин, карнитин не выходят из мышечной клетки. Проницаемость мембран существенно меняется при некоторых фармакологических нагрузках, гормональных воздействиях. Трудно объяснить максимальный уровень гиперферментемии при болезни Дюшенна в доклинической стадии при отсутствии еще некроза мышечной ткани, а также механизм гибели мышечных волокон при доброкачественных формах миодистрофии, где степень ферментемии и креатинурии очень незначительна.Гипотеза нарушения обмена циклических нуклеотидов. Циклические нуклеотиды (циклический аденозинмонофосфат -ц. АМФ, циклический гуанинмонофосфат - ц. ГМФ) играют ведущую роль в процессах метаболизма мышечного волокна, ц. АМФ контролирует активность ключевых ферментов углеводного и липидного обмена, кальцийсвязывающую способность саркоплазматического ретикулума, работу генетического и белково-синтетического аппарата, проницаемость сарколеммы и лизосомальных мембран. Свое регулирующее влияние на метаболизм внутри клетки п. АМФ осуществляет через систему протеинкиназ. Основные биохимические признаки дистрофического процесса (эмбриональные черты метаболизма, накопление жира, усиление протеолиза, переход веществ в кровяное русло) таким образом могут быть объяснены нарушением обмена циклических нуклеотидов.Уровень ц. АМФ зависит от активности его ферментов- встроенной в мембрану (связанную с бета-адренорецепторами) аденилатциклазы, катализирующей синтез, и фосфодиэстеразы, осуществляющей распад нуклеотида до неактивного АМФ. Содержание ц. АМФ можно изменить, вводя ингибиторы и активаторы этих ферментов. Так, метилксантины, цитрат натрия, тормозя активность фосфодиэстеразы, увеличивают концентрацию нуклеотида. Тот же эффект может быть получен введением адреналина и фторида натрия, стимулирующих аденилатциклазу. Бета-адреноблокаторы (пропранолол) и активаторы фосфодиэстеразы (имидазол) снижают уровень ц. АМФ.Немногочисленные данные, имеющиеся в литературе, говорят о том, что у больных миодистрофией Дюшенна ослаблены реакции, находящиеся под контролем циклических нуклеотидов.Искусственное увеличение уровня ц. АМФ при назначении больным с болезнью Дюшенна метилксантинов в субмаксимальных суточных дозах уже через несколько часов приводит к резкому снижению ферментемии, креатинурии и аминоацидурии, а также улучшению общего состояния больного. Дополнительная блокада бета-адренорецепторов (например, при введении пропранолола) у этих больных дает обратные биохимические сдвиги и приводит к ухудшению самочувствия, увеличению мышечной слабости.У больных с миодистрофией Эрба и Ландузи-Дежерина установлен противоположный характер метаболических сдвигов по сравнению с Х-сцепленными формами миодистрофии. Так, 10-дневный курс лечения анаприлином приводит к закономерному уменьшению креатинурии в среднем на 40%, аминоацидурии на 50%, активности КФК более чем в 1.5 раза[Полякова Н. Ф„ 1978].Полученные данные о важной роли ц. АМФ в развитии дистрофического процесса показали таким образом различный характер биохимических сдвигов при разных формах мышечных дистрофий. Они послужили основой для разработки принципиально нового метода лечения миодистрофии Эрба и Ландузи - Дежерина с применением бета-адреноблокаторов. Остается недостаточно доказанной первичность выявленных изменений обмена циклических нуклеотидов.Лечение при первичных мышечных дистрофиях
Отсутствие данных о первичном биохимическом дефекте и патогенезе заболевания затрудняет проведение рациональной терапии. Накопленный опыт говорит о том, что систематическое проведение комплексных курсов лечения в ряде случаев способствует некоторому замедлению патологического процесса, иногда и его стабилизации.Во все комплексы должны включаться ЛФК и массаж, которые способствуют поддержанию мышечного тонуса, улучшают периферическое кровоснабжение, задерживают развитие контрактур. Важное место отводится дыхательной гимнастике. Аналогичный принцип лежит в основе рекомендаций назначения сосудорасширяющих препаратов в сочетании с оксигенотерапией, физиотерапией, бальнеотерапией (радоновые или сульфидные ванны). Следует иметь в виду, что физиотерапия и особенно бальнеотерапия рекомендуются только в ранних стадиях процесса или при доброкачественных, медленно прогрессирующих формах миодистрофий.Назначение анаболических гормонов должно проводиться с большой осторожностью, короткими курсами (ретаболил 1 раз в 5-7 дней, на курс лечения 5-6 инъекций) с одновременным назначением переливания одногруппной крови по 100-150 мл. Прямым показанием для введения этой группы препаратов является гипогонадизм у лиц мужского пола.Могут быть рекомендованы витамин Е внутрь или внутримышечно (инъекции эревита), витамины группы В, никотиновая кислота. Показано лечение монокальциевой солью АТФ по 3-6 мл в день внутримышечно в течение месяца.Проводят лечение аминокислотами (гликокол, лейцин, глутаминовая кислота) и оротатом калия.Неврологи в Москве
Неврологи в Москвеvse-zabolevaniya.ru
Мышечная дистрофия - причины, симптомы и лечение
Вы здесь: Мышечная дистрофия У вас в браузере отключен java script, вам надо его включить или вы не сможете получить всю информацию по статье «Мышечная дистрофия и симптомы проявления». Категория: Суставы, кости, мышцы Просмотров: 27651Мышечная дистрофия - основные симптомы:
- Слабость в ногах
- Атрофия мышц
- Усталость
- Отсутствие боли
- Снижение мышечного тонуса
- Изменение походки
- Изменение размеров мышц
- Утрата физических навыков
Мышечная дистрофия является группой хронических заболеваний мышечных структур, преимущественно скелетных. Для всех прогрессирующих мышечных дистрофий характерной чертой является постепенно проявляющаяся слабость мышц, а также их дегенерация. По мере развития недуга наблюдается уменьшение диаметра мышечных волокон. Поражённые элементы в результате дистрофии утрачивают свою способность сокращаться и постепенно распадаются. Их место в теле больного человека занимает соединительная и жировая ткань.
Клиницисты выделяют всего девять разновидностей этого патологического состояния, которые имеют существенные различия в зависимости от агрессивности развития, основных характеристик, локализации поражённых волокон, а также возрастных показателей.
Этиология
Пока ещё учёным точно не удалось выяснить истинные причины, которые запускают патологические механизмы, провоцирующие патологию. Но точно уже известно, что основа всех причин этого патологического состояния – мутации аутосомно-доминантного генома, основной функцией которого является синтезирование и регенерация в теле человека специфического белка, отвечающего за полноценное формирование мышечных волокон.
В зависимости от того, какая именно хромосома в генетическом коде человека была подвержена процессу мутации, зависит, недуг с каким расположением будет развиваться:
- в большинстве клинических ситуаций мутации подвергается Х хромосома в геноме человека. В этом случае начинает прогрессировать мышечная дистрофия Дюшена. Именно эта форма недуга диагностируется чаще всего. Примечателен тот факт, что если представительница прекрасного пола имеет в своём геноме дефектную хромосому, то велика вероятность того, что она передаст её и своим потомкам. Но бывает и так, что у неё никаких симптомов недуга проявляться и вовсе не будет;
- причиной мотонической разновидности недуга является формирование аномального генома, относящегося к 19 хромосоме;
- отдельно стоит выделить мышечную недоразвитость, которая совершенно не связана с аномалиями в половой хромосоме. В данную группу относят 2 вида болезни: плечо-лопатка-лицо, поясница-конечности.
Разновидности
Клиницисты выделяют несколько наиболее распространённых форм недуга.
Мышечная дистрофия Дюшена. Данную разновидность также именуют в медицинской литературе псевдогипертрофической. Обычно мышечная дистрофия Дюшена начинает прогрессировать уже в детском возрасте. Примечателен тот факт, что этому недугу более подвержены маленькие мальчики, нежели девочки.
Симптомы мышечной дистрофии Дюшена проявляются у малышей уже в возрасте от 2 до 5 лет. Изначально поражаются мышцы ног, тазового пояса. Но постепенно патологический процесс «перебирается» на мышцы верхней части туловища. Позже вовлекаются и остальные мышечные структуры. Примечателен тот факт, что недуг стремительно прогрессирует, и уже в среднем к 12–15 годам больной полностью утрачивает способность свободно передвигаться. Прогноз этого типа дистрофии не является благоприятным – многие люди погибают, даже не достигнув 20-летнего возраста.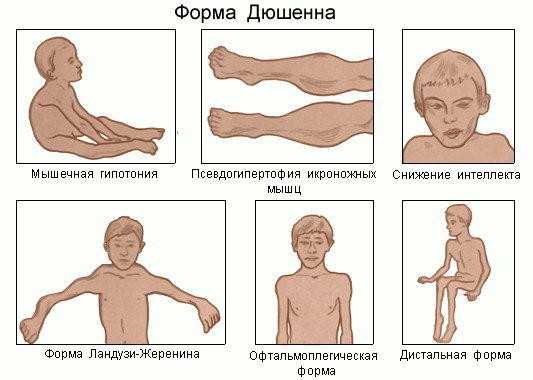
Прогрессирующая мышечная дистрофия Дюшена
Болезнь Штейнерта. Эта разновидность патологии является характерной для взрослых людей из возрастной категории от 20 до 40 лет. Редки случаи, когда патология проявляет себя уже в младенческом возрасте. Ограничений, касательно половой принадлежности, дистрофия не имеет. Характеризуется медленным прогрессированием.
Дистрофия этого вида имеет свою характерную черту – патологический процесс затрагивает не только мышцы скелета, но также и структуры жизненно важных органов. У больного возможно появление слабости мышц лица. Стоит отметить, что поражение других групп мышц также не исключено. Характерно медленное расслабление волокон после их предварительного сокращения.
Прогрессирующая мышечная дистрофия Беккера. Эта разновидность патологии является малораспространенной. Патологический процесс прогрессирует довольно медленно. Прогрессирующую мышечную дистрофию Беккера обычно диагностируют у людей с низким ростом. Прогноз недуга благоприятный. На протяжении многих лет люди с таким диагнозом сохраняют свою работоспособность, и их состояние остаётся удовлетворительным. Инвалидизации способствуют травмы различной степени тяжести, а также сопутствующие болезни.
Юношеская мышечная дистрофия Эрба-Рота. Период проявления её симптомов – от 10 до 20 лет. Прогрессирует медленно. На начальных стадиях развития отмечается атрофия волокон рук и плеч, позже – ног и таза. Во время ходьбы можно отметить изменение осанки человека – грудная клетка отодвигается немного назад, в то время как живот выпячивается вперёд. Больной идёт и переваливается.
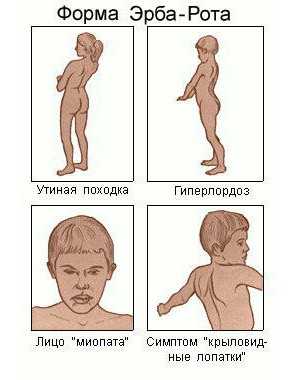
Мышечная дистрофия Эрба-Рота
Мышечная дистрофия Ландузи-Дежерина. Симптомы недуга проявляются в период от 6 до 52 лет. Чаше всего признаки дистрофии Ландузи-Дежерина выявляют в период с 10 до 15 лет. При этом недуге в первую очередь страдают мышцы лица. Но постепенно при дистрофии Ландузи-Дежерина патологический процесс охватывает также мышечные структуры конечностей и туловища.
Первым симптомом развития болезни является неполное смыкание век. Постепенно перестают полностью смыкаться губы, что обуславливает нарушение дикции. Патология Ландузи-Дежерина у больных протекает достаточно медленно. У пациента длительное время сохраняется способность двигаться, поэтому он может вести нормальный образ жизни. В среднем через 20–25 лет возможно атрофирование мышц тазового пояса, что и становится причиной инвалидизации. В целом можно сказать, что дистрофия Ландузи-Дежерина протекает благоприятно.
Симптоматика
Различные формы дистрофии (Ландузи-Дежерина, Дюшена, Беккера и прочее) имеют свои характерные признаки прогрессирования. Но также существует группа симптомов, которые являются характерными для любой разновидности патологии:
- отсутствие болевого синдрома;
- постепенное понижение тонуса мышечных волокон;
- чувствительность в поражённых участках не понижается;
- скелетные мышцы постепенно атрофируются;
- перемена походки;
- частые падения вследствие слабости мышц ног;
- постоянная усталость;
- характерный симптом прогрессирования недуга – изменение размеров мышц;
- мышечная дистрофия у детей проявляется постепенным утрачиванием физ. навыков, которые они уже успели развить до момента прогрессирования патологии.
Диагностика
Диагностика мышечной дистрофии у детей и взрослых включает в себя такие мероприятия:
- тщательный сбор анамнеза;
- электромиография;
- взятие небольшого участка мышечного волокна для проведения микроскопического исследования;
- дополнительное консультирование у ортопеда и терапевта.
Осложнения
- нарушения работы сердца;
- деформация позвоночного столба;
- снижение интеллекта, функций памяти;
- снижение способность совершать активные движения и постепенна инвалидизация;
- прогрессирование патологий системы дыхания;
- летальный исход (медицинская статистика такова, что этот недуг редко становится причиной смерти).
Лечебные мероприятия
Стоит сразу отметить, что мышечную дистрофию излечить полностью возможности нет. Ещё не было создано такого препарата или процедуры, которые смогли бы восстановить поражённые участки мышечных волокон. Лечение мышечной дистрофии в первую очередь направлено на торможение активного развития процессов дистрофии в мышечных структурах. С этой целью больному назначают кортикостероиды, витамины, АТФ и прочее.
Дополнительно назначают:
- лечебный массаж;
- физиотерапию;
- дыхательную гимнастику;
- проводят профилактику прогрессирования сколиоза.
Поделиться статьей:
Если Вы считаете, что у вас Мышечная дистрофия и характерные для этого заболевания симптомы, то вам могут помочь врачи: ортопед, хирург, невролог.
Также предлагаем воспользоваться нашим сервисом диагностики заболеваний онлайн, который на основе введенных симптомов подбирает вероятные заболевания.
Заболевания со схожими симптомами:
Атаксия Фридрейха — генетическая патология, при которой отмечается поражение не только нервной системы, но и развитие экстраневральных нарушений. Болезнь считается довольно распространенной — с таким диагнозом живет 2–7 человек на 100 тысяч населения.
... Миозит (совпадающих симптомов: 2 из 8)Поражение мышц, обусловленное травматическим, воспалительным или токсическим характером и возникающее вследствие влияния различных факторов преимущественно на мышечные волокна, вызывая их ослабление и даже атрофию, называется миозитом. Он представляет собой заболевание, которое отображается преимущественно на скелетных мышцах человека: спине, шее, грудной клетке и прочих группах.
...Ревматическая полимиалгия – воспалительное заболевание, которое проявляется в виде болей в мышцах плечевого и тазового пояса, которые часто могут сопровождаться лихорадкой и значительной потерей веса. Точная этиология патологии до сих пор неизвестна. К общей клинической картине может прибавиться симптоматика темпорального артрита. Больше всего заболеванию подвержены люди от 50 до 75 лет. Женщины страдают от этого недуга гораздо чаще, чем мужчины.
...Боковой амиотрофический склероз – недуг нейродегенеративного характера, при котором поражаются периферические, а также центральные мотонейроны (двигательные нервные волокна). Вследствие прогрессирования этого синдрома у больного человека наблюдается атрофия скелетной мускулатуры, фасцикуляции, гиперрефлексия и прочие расстройства. Остановить протекание патологии на это время не является возможным, поэтому постепенное усиление симптоматики неизбежно приводит к летальному исходу.
...Миопатия – врождённая патология, причиной которой являются определённые мутации в генах. Механизм развития болезни окончательно не изучен, поэтому врачи не могут точно определить, когда у пары может родиться больной ребёнок. Бывает и так, что у совершенно здоровых отца и матери может появиться ребёнок с любой формой миопатии. Вообще же болезнь связана с нарушениями обменных процессов в тканях мышц, которые из-за этого теряют креатин, что приводит к их дистрофии.
...simptomer.ru
причины заболевания и лечение, симптомы дистрофии мышц
 Мышечная дистрофия (МД) — это группа заболеваний, характеризующихся прогрессирующей слабостью и мышечной дегенерацией. Мышцы постепенно атрофируются — теряют свой объем и, следовательно, силу.
Мышечная дистрофия (МД) — это группа заболеваний, характеризующихся прогрессирующей слабостью и мышечной дегенерацией. Мышцы постепенно атрофируются — теряют свой объем и, следовательно, силу.
Это болезни генетического происхождения, которые могут возникать в любом возрасте: с самого рождения, в детстве или во взрослой жизни. Существует более 30 форм заболеваний, которые различаются по возрасту появления симптомов, характеру пораженных мышц и степени тяжести. Большинство типов дистрофий постепенно осложняются и имеют необратимые последствия. В настоящее время лечения МД все еще не существует. Наиболее известным и распространенным типом заболеваний является миопатия Дюшенна.
В ходе развития МД страдают в первую очередь основные мышцы, которые способствуют произвольному движению, включая мышцы, бедра, ног, рук и предплечья. В некоторых случаях могут быть затронуты респираторные мышцы и сердце. Люди с мускульной дистрофией постепенно теряют свою мобильность при ходьбе. Другие симптомы могут быть связаны с мышечной слабостью, включая сердце, желудочно-кишечные, глазные проблемы.
Также рекомендуем прочитать:
Дистрофия или миопатия? Термин «миопатия» является общим названием для всех патологий М. Д. Мышечные дистрофии — это особые формы миопатий. Однако в повседневном языке термин миопатия часто используется для обозначения дегенерации мышц.
Содержание материала
Распространенность заболевания
Миопатия относится к редким и неизлечимым заболеваниям. Трудно вывести точную статистику, поскольку она объединяет различные болезни. Согласно некоторым исследованиям, около 1 из 3 500 человек страдают от этого заболевания.
Например:
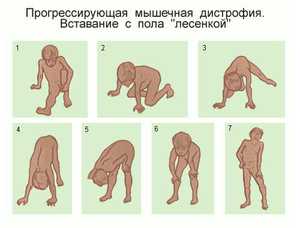 Миопатия Дюшенна затрагивает приблизительно одного ребенка (мальчика) из 3500.
Миопатия Дюшенна затрагивает приблизительно одного ребенка (мальчика) из 3500.- Миопатия Беккера касается 1 мальчика из 18 000.
- Фазио-скапулогумаральная дистрофия поражает около 1 из 20 000 взрослых людей.
- Болезнь Эмери-Дрейфус затрагивает 1 из 300 000 человек, вызывает ретракцию сухожилия и нарушение сердечной мышцы
Частота и тип заболеваний зависит от конкретной страны:
 Врожденная миопатия, известная как Фукуяма, распространена в основном Японии.
Врожденная миопатия, известная как Фукуяма, распространена в основном Японии.- В Квебеке доминирует окулофарингеальная дистрофия (1 случай на 1000 человек), тогда как в остальном мире это очень редкое заболевание (1 случай на 100 000 в среднем). Как следует из названия, эта патология в основном касается мышц век и горла.
- Болезнь Штейнтера или «миотония Штайнерт» широко распространена в регионе Сагеней-Лак-Сен-Жан — затрагивает около 1 из 500 человек.
- Саркогликанопатия более распространена в Северной Африке и поражает одного человека из 200,000 на северо-востоке Италии.
- Кальпайнопатия была впервые описана на острове Реюньон. Затрагивает одного из 200 000 человек.
Причины заболевания и лечение
Причиной данной патологии являются генетические заболевания, то есть дефект (или мутация) гена, необходимого для нормального развития мышц. Когда этот ген мутирует, мышцы больше не в состоянии нормально функционировать — они теряют свой силовой потенциал и в результате атрофируются.
В ходе миопатии участвует несколько десятков разных генов. Чаще всего это гены, «производящие» белки, которые расположены в мембране мышечных клеток.
Например:
- Миопатия Дюшенна связана с дефицитом дистрофина — белка, расположенного под мембраной мышечных клеток, который играет роль в сокращении мышц.
- Почти у половины врожденных МД причиной является дефицит мерозина — белка, составляющего мембрану клеток мышцы.
 Как и многие генетические заболевания, миопатия чаще всего передается родителями их ребенку. Реже эти заболевания могут «появляются» спонтанно, когда ген мутирует случайно. В этом случае больной ген отсутствует у родителей или других членов семьи.
Как и многие генетические заболевания, миопатия чаще всего передается родителями их ребенку. Реже эти заболевания могут «появляются» спонтанно, когда ген мутирует случайно. В этом случае больной ген отсутствует у родителей или других членов семьи.
Как правило, МД передается рецессивно. Другими словами, для того чтобы болезнь выражалась, оба родителя должны быть носителями и передавать ребенку ненормальный ген. Болезнь не проявляется у родителей по той причине, что у каждого из них есть только один аномальный ген, а не два. Для нормального функционирования мышц достаточно одного нормального гена.
Кроме того, некоторые формы миопатии затрагивают только мальчиков: это миопатия Дюшенна и Беккера. В обоих случаях ген, участвующий в этих двух заболеваниях, расположен в Х-хромосоме, которая существует в единственной копии у мужского пола.
Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Характеристики наиболее распространенных патологий
 Мышечная миопатия Дюшенна. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Мышечная миопатия Дюшенна. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
Миопатия Беккера. Симптомы сравнимы с симптомами М. Д. Дюшенна , однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Также наблюдается Миотония — аномальное и продолжительное сокращение мышц (мышца расслабляется слишком медленно), особенно выражается в руках, а иногда и на языке. Также могут быть затронуты мышцы лица, шеи и лодыжек. Часто присутствуют сердечные и дыхательные нарушения, которые являются потенциально серьезными. Нередко наблюдаются пищеварительные, гормональные, глазные расстройства, а также бесплодие и раннее облысение.
Миопатия поясничного отдела. Симптомы обычно проявляются в детстве (10 лет) или в раннем взрослом возрасте (около 20 лет). Мышцы плеч и бедер постепенно ослабевают, в то время как мышцы головы, шеи и диафрагмы обычно не затрагиваются. Если некоторые формы сопровождаются дыхательными нарушениями, то при этом типе дистрофии такие аномалии отсутствуют. Сердечные нарушения встречаются редко. Эволюция (развитие заболевания) очень изменчива, в зависимости от формы.
Миопатия Дежерина-Ландузи или плечелопаточная дистрофия. Симптомы обычно появляются в позднем детстве или в зрелом возрасте (от 10 до 40 лет). Как следует из названия, миопатия затрагивает мышцы лица, плеч и рук. Таким образом, больному становится сложно выразить улыбку, произнести некоторые предложения и закрыть глаза. Потеря подвижности происходит примерно в 20% случаев. Заболевание развивается медленно, продолжительность жизни нормальная.
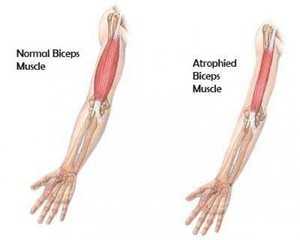 Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Окуло-глоточная миотония. Это заболевание относительно распространено в Квебеке. Симптомы обычно появляются около 40 или 50 лет. Первые признаки болезни проявляются опустившимися веками, за которыми следуют слабость мышц глаз, лица и горла (глотки), вызывая трудности с глотанием пищи. Прогрессирование заболевания происходит медленно.
Исследования и прогресс
С 2005 года для лечения пациентов с развивающимся поражением мышц все чаще используются стволовые клетки. Для лечения мышечной дистрофии этим методом могут быть рассмотрены различные варианты заболевания, такие как: мышечные дистрофии Дюшенна, Беккера, миопатия поясничного и плечевого отдела.
Целью лечения является регенерация потерянных и поврежденных мышечных волокон с использованием регенеративного потенциала стволовых клеток. Для этого большое количество стволовых клеток вводится при помощи нескольких внутривенных и внутримышечных инъекций, что позволяет лучше нацеливать терапию именно на пораженную группу мышц.
Возможный прогресс
Терапия с применением стволовых клеток может обеспечить улучшение в плане мышечной массы, силы, движений, баланса, тремора и ригидности мышц. Стволовые клетки также могут замедлить будущую потерю мышечного объема и уменьшить симптомы.
Важно отметить, что лечение не является окончательным лекарством от этого заболевания и никоим образом не может решить проблему потери мышечных волокон. По этой причине прогресс после такого лечения не может быть постоянным. Исследования в этой области все еще ведутся.
Семейства заболевания
Обычно существуют два основных семейства МД:
- Мышечна врожденная дистрофия (ВМД), которая выражается в первые 6 месяцев жизни, сопровождает около десяти форм патологий переменной тяжести, включая ВМД с первичной недостаточностью мерозина, синдром Ульриха и Уокера-Варбурга;
- Мышечные дистрофии, появляющиеся в детстве или в зрелом возрасте:
- Миопатия Дюшенна
- Миопатия Беккера
- Миопатия Эмери-Дрейфуса (существует несколько форм)
- миопатия Ландузи-Дежерина
- Так называемая миопатия поясничного отдела — затрагивает мышцы вокруг плеч и бедер.
- Миотонические дистрофии (типы I и II), которые включают болезнь Штейнтера. Они характеризуются миотонией — когда мышцы не могут нормально расслабиться после сокращения.
- Окулофарингеальная миопатия
Эволюция дистрофии
Эволюция (развитие заболевания) МД сильно варьируется от одной формы к другой, а также от одного человека к другому. Некоторые формы быстро развиваются, что приводит к ранней утрате подвижности и ходьбе, а иногда и к смертельным сердечным или респираторным осложнениям, в то время как другие развиваются очень медленно — в течение десятилетий. Большинство врожденных мышечных дистрофий, например, которые мало выражены или почти незаметны, позже могут проявятся внезапно и с серьезными последствиями.
Возможные осложнения
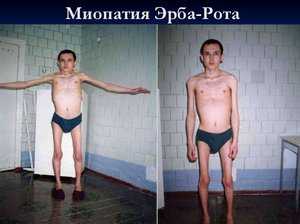 Осложнения сильно различаются в зависимости от типа патологии. Некоторые нарушения могут затрагивать респираторные мышцы или сердце, иногда с очень тяжелыми последствиями.
Осложнения сильно различаются в зависимости от типа патологии. Некоторые нарушения могут затрагивать респираторные мышцы или сердце, иногда с очень тяжелыми последствиями.
Таким образом, сердечные осложнения довольно распространены, особенно у мальчиков с мышечной дистрофией Дюшенна.
Кроме того, дегенерация мышц заставляет тело и суставы деформироваться постепенно: на фоне этого у больных может развиваться сколиоз. Часто наблюдается сокращение мышц и сухожилий, что приводит к их стягиванию. Все эти нарушения приводят к деформации суставов: ноги и руки повернуты внутрь и вниз, деформируются колени или локти.
Также известно, что болезнь сопровождается тревожными или депрессивными расстройствами, поэтому больным требуется много внимания и поддержки, в первую очередь со стороны близких.
vitaminki.guru
Прогрессирующая мышечная дистрофия лечение и ортопедические меры
Эта форма была описана врачом по имени Беккер в полвека назад. Прогрессирующая мышечная дистрофия наследуется по типу рецессивному сцепленному с Х-хромосомой. Частота наследования до сих пор не установлена. Прогрессирующая мышечная дистрофия, лечение и ортопедические меры в борьбе с болезнью - тема этой статьи.
Прогрессирующая мышечная дистрофия - признаки болезни
Первыми признаками прогрессирующей мышечной дистрофии являются:
1.
мышечная ослабленность;
2.
патологическая мышечная утомляемость, возникающая при физических нагрузках;
3.
псевдогипертрофия икроножной мышцы.
Эти признаки могут проявиться уже в десяти - пятнадцатилетнем возрасте, а иногда даже раньше. Первые симптомы прогрессирующей мышечной дистрофии, как правило, развиваются симметрично - сперва они локализуются в группах мышц, которые называются проксимальными и расположены в нижних конечностях - тазовом поясе и бедрах. Известно, что в дальнейшем эти пороки распространяются также на проксимальные группы мышц, которые расположены в верхних конечностях. Из-за атрофии могут возникнуть изменения походки, она становится как бы «утиной», возникают компенсаторные приемы в процессе вставания. При мышечной дистрофии в проксимальных группах мышц мышечный тонус умеренно снижен. Наиболее глубокие рефлексы многих мышц какое-то время сохраняются, заметно снижаются лишь коленные рефлексы. Также умеренно выражены сердечнососудистые расстройства. Порой при прогрессирующей мышечной дистрофии наблюдаются болезненные ощущения в области сердца, возникает блокада ножек в пучке Гиса. Проявляются эндокринные нарушения, которые характеризуются гинекомастией, затуханием либидо, а то и вовсе импотенцией. Однако, интеллект при этом недуге сохраняется.
Прогрессирующая мышечная дистрофия характеризуется медленно прогрессирующим течением. Темпы распространения атрофии мышц невысоки и заболевшие долгое время могут сохранять работоспособность.
Прогрессирующая мышечная дистрофия - диагностика болезни
Диагноз может быть поставлен на основании специального генеалогического анализа, на основании данных биохимических исследований, а также биопсии мышц, проявляющих первичный мышечный тип изменений.
Дифференцировать эту болезнь нужно с такими прогрессирующими мышечными дистрофиями, как болезнь Дюшена, а также заболевание Эрба-Рота, а кроме того – с известным заболеванием Кугельберга-Веландер.
Сегодня причина этого заболевания остается неизвестной. Понятно лишь, что оно часто бывает семейным и передается по наследству. При его возникновении фиксируется прогрессирующая гибель волокон мышц с заменой их соединительной тканью.
Прогрессирующая мышечная дистрофия - лечение болезни
Главная задача врача в этом случае состоит в том, чтобы по возможности дольше затянуть период, в течение которого человек еще способен передвигаться самостоятельно, поскольку в лежачем положении стремительно нарастают контрактуры, а вместе с ними приходит сколиоз и разнообразные дыхательные расстройства. Типичный комплекс при лечении прогрессирующей мышечной дистрофии обязан включать такие средства:
1.
лечебная гимнастика;
2.
массаж;
3.
различные ортопедические мероприятия;
4.
медикаментозная терапия.
Прогрессирующая мышечная дистрофия и лечебная гимнастика
Лечебная гимнастика должна состоять не только из пассивных, но также и активных движений, которые выполняются больным для всех суставов в различных положениях:
1.
стоя
2.
сидя
3.
лежа
4.
при разном положении конечностей тела.
Динамичные движения желательно выполнять в режиме, который называется изометрическим. Занятия подобной гимнастикой при лечении прогрессирующей мышечной дистрофии лучше всего проводить регулярно, делая в день несколько подходов. В это же время требуется предостеречь новичка от чрезмерных перенапряжений, особенно таких, которые сопровождаются растяжением групп мышц. Большое значение имеют разнообразные дыхательные упражнения.
Прогрессирующая мышечная дистрофия и ортопедические меры
Мышечная дистрофия, лечение которой предполагает ортопедические мероприятия, протекает существенно медленнее. Этому способствует консервативный и оперативный характер мероприятий. При этом в любом случае требуется индивидуально определить предполагаемую пользу и риск вреда от столь оперативного и настойчивого вмешательства. Нужно, кроме того, учитывать, что часто (обычно, при ярко выраженном гиперлордозе или ослаблении четырехглавой мышцы) установка стоп эквиноварусного характера имеет важное компенсаторное значение. Поэтому после проведения, к примеру, ахиллотомии, любой пациент может оказаться совершенно обездвиженным. При развитии контрактур специалисты рекомендуют проводить бережное и осторожное растягивание мышц около 30 раз в день с наложением на них шины в период сна.
www.astromeridian.ru