Синдром Прадера-Вилли: описание, диагностика, фото, лечение. Синдром прадера вилли фото
Синдром Прадера - Вилли | EUROLAB
Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г.
По данным регистра ассоциации больных с синдромом Прадера-Вилли, в США и Канаде на декабрь 1986 г. насчитывалось 1595 больных. В последние годы удалось установить популяционную частоту патологии, составляющую 1 : 10 000 - 1 : 20 000.
Авторы, впервые описавшие синдром, высказывали предположение об аутосомно-рецессивном типе наследования заболевания. Затем появились сообщения о возможности аутосомно-доминантной передачи болезни. Подтверждением данных гипотез могли служить наблюдавшиеся семейные случаи патологии. Однако большинство описанных клинических наблюдений синдрома Прадера - Вилли носило спорадический характер.
Последующие исследования позволили установить у детей с синдромом Прадера - Вилли определенные хромосомные нарушения. Цитогенетический анализ показал, что хромосомные аномалии у больных были представлены либо транслокациями (t 15/15), либо мозаицизмом. В 1987 г. появились первые сообщения о микроделеции хромосомы 15. Однако окончательная идентификация хромосомных изменений при синдроме Прадера - Вилли стала возможной только после внедрения в практику молекулярно-генетических методов исследования.
В настоящее время установлено, что развитие синдрома Прадера - Вилли связано с повреждением критического района хромосомы 15 (сегмента q11.2- q13). При этом оказалось, что повреждение этого же участка хромосомы 15 наблюдается и при другом заболевании - синдроме Ангельмана, клиническая картина которого существенно отличается от синдрома Прадера - Вилли и характеризуется ранним (в возрасте 6-12 мес) замедлением психомоторного развития, микроцефалией, нарушением речи (в 100% случаев), атаксией, неконтролируемым насильственным смехом, частыми эпилептиформными припадками, специфическим выражением лица.
Таким образом, несмотря на повреждение при синдромах Прадера - Вилли и Ангельмана одного и того же локуса хромосомы 15, клинические проявления обеих болезней резко противоположны.
Объяснение фенотипических различий получено лишь в последние годы. Оказалось, что развитие этих заболеваний связано с новыми генетическими явлениями - геномным импринтингом и унипарентальной дисомией.
Геномный импринтинг - новое явление, открытое благодаря успехам молекулярной генетики. Он означает различную экспрессию генетического материала (гомологичных аллелей) в хромосомах в зависимости от отцовского или материнского происхождения, т.е. свидетельствует о влиянии родителей на фенотип ребенка. До настоящего времени считалось, что вклад в проявляемость (экспрессию) генов отца и матери равноценен.
По сути геномный импринтинг - это половой и тканевозависимый сложный модификатор генной активности некоторых локусов хромосом в зависимости от их родительского происхождения. Проявления геномного импринтинга выявлены и при других заболеваниях - синдромах Сотоса, Беквита-Видемана, Сильвера-Рассела, муковисцидозе и других.
Унипарентальная (однородительская) дисомия - наследование обеих хромосом только от одного из родителей. В течение многих лет считалось, что такое наследование невозможно. Лишь с помощью молекулярно-генетических маркеров удалось доказать возможность однородительской дисомии. Природа унипарентальной дисомии окончательно не выяснена, однако установлено, что она обязана своим происхождением ряду генетических и биохимических нарушений.
Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить микроделецию или унипарентальную дисомию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы - прометафазный анализ, использование ДНК-маркеров определенных участков хромосомы 15 (исследование процессов метилирования) и др.
На сегодняшний день синдромы Прадера - Вилли и Ангельмана служат общепринятой моделью для изучения новых в клинической генетике и сложных явлений - геномного импринтинга и унипарентальной дисомии.
Установлено, что синдром Прадера - Вилли может быть обусловлен двумя основными механизмами. Первый из них - микроделеция хромосомы 15 (15q11.2-q13), которая всегда отцовского происхождения. Второй - материнская изодисомия, т.е. когда обе хромосомы 15 получены от матери. Развитие синдрома Ангельмана, наоборот, связано с микроделецией того же участка хромосомы 15, но материнского происхождения, или отцовской изодисомией. Большинство (около 70%) случаев синдрома Прадера - Вилли обусловлено микроделецией, остальные - дисомией. При этом обращает на себя внимание отсутствие клинических различий между больными с микроделецией и изодисомией.
Характеристика аномалий
Дети с синдромом Прадера - Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание.
В течение заболевания можно выделить две фазы: первая - свойственна детям 12-18 мес жизни. Она характеризуется выраженной мышечной гипотонией, снижением рефлексов - Моро, сосательного и глотательного, что затрудняет кормление ребенка. Вторая - наступает позже, через несколько недель или месяцев. Появляются полифагия, постоянное чувство голода, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей.
Рост больных нередко снижен. У 75% детей наблюдается гипопигментация кожи, волос и радужки. Часто диагностируется микроцефалия. Психомоторное развитие отстает от возрастной нормы - коэффициент интеллектуального развития - от 20 до 80 ед. (при норме 85-115 ед.). Речь затруднена, словарный запас уменьшен. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, страбизм.
Встречаются и другие аномалии: микродонтия, гипоплазия хрящей ушных раковин, сколиоз, эктропион (выворот века), глаукома.
Нередко развитие сахарного диабета, который с возрастом имеет тенденцию к улучшению.
При морфологическом исследовании мозга и ЯМР-томографии могут наблюдаться (примерно в 12% случаев) кисты червя мозжечка, аномалии коры головного мозга.
Продолжительность жизни больных может достигать 60 лет и более.
Патогенез
Патогенез синдрома Прадера - Вилли до настоящего времени остается малоисследованным. Высказываются предположения, что ожирение у больных обусловлено значительным (более чем в 10 раз) усилением синтеза жира из ацетата и крайне низкими процессами липолиза.
Гипогонадизм по гипогонадотропному типу может быть связан с дисфункцией гипоталамуса, преимущественно, в области вентромедиального и вентролатерального ядер. Правильность данной точки зрения подтверждается эффективностью лечения больных фармацевтическими препаратами (кломифен), приводившими к увеличению в плазме содержания лютеинизирующего гормона, тестостерона, нормализации показателей почечной экскреции гонадотропинов, сперматогенеза и появлению вторичных половых признаков.
Одним из объяснений гипопигментации кожи, волос и радужки служит снижение активности тирозиназы в волосяных фолликулах и меланоцитах, а также уменьшение пигмента в сетчатке.
Обращается внимание на повышенный риск развития лейкемии у больных с синдромом Прадера - Вилли. Исследования выявили снижение репарации ДНК (до 65% по сравнению с 97% у здорового ребенка) в лимфоцитах больных с данной патологией. Не исключено, что низкая репарационная способность ДНК может играть роковую роль в развитии злокачественных новообразований у лиц с синдромом Прадера - Вилли.
Лечение
Терапия синдрома Прадера - Вилли окончательно не разработана. По данным литературы, комплекс лечебных мероприятий включает лишь диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины).
www.eurolab.ua
Синдром Прадера-Вилли - диганостика, лечение, симптомы
Синдром Прадера-Вилли является редкой наследственной болезнью, возникающей из-за отсутствия отцовской копии одного из участков хромосомы. Впервые он описан в середине ХХ века швейцарскими учеными А. Прадером и Х. Вилли. Заболевание встречается у 1:12000-15000 рожденных детей.
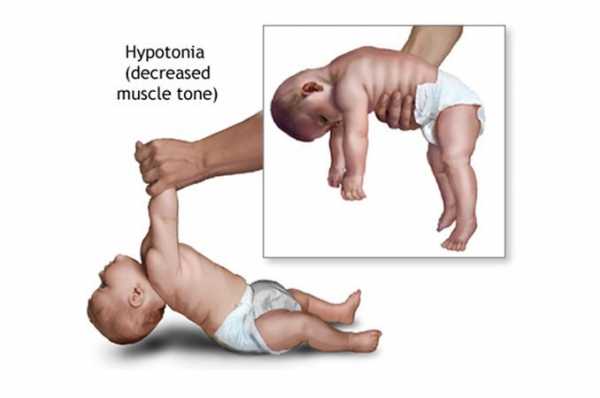
Клиническая характеристика синдрома Прадера-Вилли
Дети с этой наследственной болезнью обычно рождаются доношенными. Часто у них наблюдается нерезко выраженная внутриутробная гипотрофия (низкая подвижность плода). Врачи различают 2 фазы синдрома Прадера-Вилли:
- Первая характеризуется выраженной мышечной гипотонией. Часто ее дополняют: рефлекс Моро, снижение сухожильных рефлексов, тенденция к гипотермии;
- Вторая фаза болезни обычно развивается через несколько месяцев и проявляется в виде полифагии. Дети не могут утолить свой голод, поэтому они готовы есть непрерывно, что приводит к развитию ожирения. При этом жир откладывается чаще всего на проксимальных отделах конечностей и на туловище, в то время как кисти и стопы диспропорционально маленькие. Во второй фазе признаки гипотонии несколько уменьшаются.
У таких детей рост обычно снижен. У мальчиков возникает гипоплазия мошонки, полового члена, крипторхизм. Девочкам характерны гипоплазия половых губ, а в более взрослом возрасте –аменорея и гипоплазия матки.
Как правило, у больных детей психомоторное развитие отстает от нормы. У них хорошая зрительная долговременная память, и они могут научиться читать, но их собственная речь обычно отстает от понимания. Математические навыки, слуховая память и навыки письма чаще всего развиты хуже, что требует особого подхода при обучении. Синдром Прадера-Вилли у детей характеризуется затрудненной речью и маленькимсловарным запасом. Как правило, больные доброжелательны по характеру, плохо умеют управлять своими эмоциями, безынициативны, и им свойственны резкие перепады настроения.
Помимо этих основных симптомов может наблюдаться:
- Высокое арковидное небо;
- Сухая слизистая полости рта;
- Микроцефалия;
- Кариес и дефекты эмали;
- Гипоплазия хрящей ушных раковин;
- Судороги и страбизм;
- Микродонтия;
- Мезобрахифалангия;
- Сколиоз;
- Клинодактилия;
- Синдактилия;
- Нарушение координации;
- Поперечная ладонная складка.
Также нередко в период совершеннолетия синдрому сопутствует сахарный диабет, аномальная гибкость и жидкие лобковые волосы. Обычно у больных встречается не больше пяти указанных признаков.
Общими внешними признаками взрослых с синдромом Прадера-Вилли являются:
- Широкий и большой нос;
- Избыточный вес с жировыми отложениями в центральной части тела;
- Чувствительная кожа, на которой легко образуются синяки и царапины;
- Маленькие ноги и руки с непропорционально узкими пальцами.

Диагностика и лечение синдрома Прадера-Вилли
Диагностика синдрома Прадера-Вилли проводится на основании симптомов болезни, которые подтверждают генетическим анализом. Такое исследование рекомендуется проводить всем детям с пониженным мышечным тонусом.
Необходимо отметить, что при помощи обычного исследования хромосомного состава кариотипа диагностировать унипарентальную дисомию или микроделецию нельзя. Для этого применяют специальные молекулярно-генетические и цитогенетические методы – прометафазный анализ, а также используют ДНК-маркеры определенных участков хромосомы 15 и некоторые другие.
Сегодня синдромы Ангельмана и Прадера-Вилли приняты за общую модель для изучения новых и сложных явлений клинической генетики – унипарентальной дисомии и геномного импринтинга.
Без проведения генетических исследований может быть установлен неправильный диагноз –синдром Дауна или миопатия. Однако опытный генетик обычно точно определяет болезнь, поскольку такие дети, как правило, имеют много схожих признаков.
После проведения диагностики синдрома Прадера-Вилли врач назначит лечение, направленное на повышение качества жизни. Обычно лечение синдрома Прадера-Вилли включает массаж и другую специальную терапию. Также рекомендованы занятия с дефектологом и логопедом и использование различных методик развития ребенка. Довольно часто применяется медикаментозная терапия, включающая прием «гормонов роста» и проведение заместительной гормональной терапии с применением гонадотропинов.
На фоне неопущения яичек и микропении у мальчиков может потребоваться хирургическое вмешательство или гормонотерапия. Для того чтобы корректировать повышенный вес, следует соблюдать диету с ограничением количества углеводов и жиров.
Поскольку синдром Прадера-Вилли – генетическая болезнь, терапия может только улучшить качество жизни больного. И чем раньше будет поставлен диагноз, тем больше шансов сохранить здоровье и создать условия для социальной адаптации больных.
zdorovi.net
симптомы, причины, лечение, профилактика, осложнения
Причины
В процессе внутриутробного развития, в хромосомном наборе ребёнка возникают нарушения. В частности, они касаются функционирования участка q11-13 пятнадцатой пары. Отвечая на вопрос от чего может быть, и как проявляется в дальнейшем рассматриваемое заболевание, специалисты всегда связывают его с нарушениями на уровне именно отцовских хромосом.
Очень часто синдром Прадера-Вилли сопровождается:
- Делецией, то есть полной утратой региона Q 11-13 гаметы отца с частотой 70 случаев из 100.
- Отсутствием копии пятнадцатой хромосомы отцовского происхождения и последующей заменой данной копии материнской с частотой 20 случаев из 100.
- Дезактивацией на стадии эмбрионального развития в следствии мутации молекулы и сохранения нуклеотидной последовательности ДНК с частотой 5 случаев из 100.
В нормальном состоянии, копия гена, полученная со стороны отца, должна работать, тогда как материнская не функционирует. Таким образом, невозможность решения естественных задач в виду отсутствия этой рабочей копии, может вызвать развитие заболевания.
Симптомы
Первые признаки болезни проявляются уже при рождении. Абсолютно большинство малышей с диагнозом синдром Прадера-Вилли появляются на свет недоношенными с очевидной внутриутробной гипотрофией, т. е., нетипично низкой подвижностью. В зависимости от фазы, заболевание характеризуется:
- Гипотонией мышц. Дополнительно синдром сочетается с рефлексом Моро, недостаточностью сухожильных рефлексов, склонностью к гипотермии.
- Полифагией. Яркие проявления заболевания в виде постоянного ощущения неутолимого голода начинаются уже с возраста 6-12-ти месяцев. Дети с синдромом Прадера-Вилли всегда имеют ожирение. Отложение подкожного жира происходит в области проксимальных частей конечностей, на торсе. Кисти ребёнка и его стопы остаются маленькими, не соответствующими пропорциям тела. Гипотония на данной фазе минимизируется.
Несовершеннолетние пациенты отстают от своих сверстников по физическому развитию. Отмечается гипоплазия в области наружных половых органов – внутренних у девочек, – и крипторхизм.
Психомоторное развитие детей с диагнозом синдром Прадера-Вилли несколько не соответствует норме. Ребёнок имеет отличную зрительную память, быстро учится читать, но речевое развитие замедленное, словарный запас ограничен. Несовершеннолетнему пациенту с трудом удается освоение математики, письма. Он плохо запоминает информацию на слух.
Список симптомов может дополняться:
- наличием высокого нёба в форме арки;
- сухостью слизистых оболочек ротовой полости;
- микроцефалией;
- повреждениями эмали зубов и кариесом;
- гипоплазией хрящевых тканей ушей;
- частыми судорогами и страбизмом;
- искривлением позвоночника;
- нарушениями координации;
- наличием поперечной наладонной складки.
У всех пациентов с синдромом Прадера-Вилли отмечается:
- широкий нос крупных размеров;
- избыточная масса тела с отложением жира в области живота;
- повышенная чувствительность кожи с мгновенным образованием синяков;
- непропорционально маленький размер рук и ног с узкими пальцами.
Диагностика синдрома Прадера-Вилли у ребёнка
Основанием для постановки диагноза являются симптомы заболевания, подтвержденные генетическими анализами. Для того, чтобы распознать заболевание используется метод молекулярно-генетического анализа или цитогенетического обследования. Кроме того, врачи работают с ДНК-маркерами конкретных зон 15-й и др. хромосом. Диагностировать заболевание без обращения к этим методам невозможно, в виду его схожести с другими – например, синдромом Дауна.
Осложнения
Чем опасен синдром Прадера-Вилли? Как правило, он не угрожает жизни, а прогноз при наличии лечения благоприятный. Средняя продолжительность жизни пациентов составляет 60 и более лет. Возможными угрозами здоровью и жизни являются:
- сахарный диабет;
- лейкемия.
Лечение
С учетом результатов диагностики врачи решают, что делать, чтобы улучшить качество жизни ребенка и способствовать его успешной адаптации в обществе. Вылечить заболевание полностью невозможно. А в целях предотвращения последствий и минимизации текущих симптомов, курс лечения включает в себя преимущественно физиотерапию – массаж, ЛФК и т. д.
Что можете сделать вы
Родители должны лечить ребёнка вместе с врачами. Только в таких условиях можно рассчитывать на действительно успешные результаты. Так, необходимо обеспечить пациенту регулярные занятия с логопедом и дефектологом, обучение в группах или индивидуальное. При назначении медикаментозного лечения необходимо соблюдать предписания врача.
Что делает врач
Распространенной практикой является назначение гормонотерапии. Лечение гормонами заключается в регулярных инъекциях «гормона роста». Также может использоваться введение гонадотропинов для восполнения недостатка их естественной выработки. Если заболевание сопровождается микропенией и неопущением тестикул у мальчиков, гормональной терапии может быть недостаточно, а вылечить симптом поможет только операция. В целях коррекции избыточного веса врач назначает ребёнку строгую низкоуглеводную диету.
Профилактика
Врожденное заболевание невозможно предотвратить. Все, что остается сделать родителям – не допустить осложнения. Чем раньше ребёнку будет назначено лечение, тем лучше будет качество его жизни в будущем. Ребёнок способен к адаптации в обществе, обучению и взаимодействию с окружающими.
Оцените материал:
спасибо, ваш голос принят
Вооружайтесь знаниями и читайте полезную информативную статью о заболевании синдром прадера-вилли у детей. Ведь быть родителями – значит, изучать всё то, что поможет сохранять градус здоровья в семье на отметке «36,6».
Узнайте, что может вызвать недуг , как его своевременно распознать. Найдите информацию о том, каковы признаки, по которым можно определить недомогание. И какие анализы помогут выявить болезнь и поставить верный диагноз.
В статье вы прочтёте всё о методах лечения такого заболевания, как синдром прадера-вилли у детей. Уточните, какой должна быть эффективная первая помощь. Чем лечить: выбрать лекарственные препараты или народные методы?
Также вы узнаете, чем может быть опасно несвоевременное лечение недуга синдром прадера-вилли у детей, и почему так важно избежать последствий. Всё о том, как предупредить синдром прадера-вилли у детей и не допустить осложнений.
А заботливые родители найдут на страницах сервиса полную информацию о симптомах заболевания синдром прадера-вилли у детей. Чем отличаются признаки болезни у детей в 1,2 и 3 года от проявлений недуга у деток в 4, 5, 6 и 7 лет? Как лучше лечить заболевание синдром прадера-вилли у детей?
Берегите здоровье близких и будьте в тонусе!
detstrana.ru
Синдром Прадера-Вилли - симптомы, лечение, диагностика
Синдром Прадера-Вилли – это генетическая редкая аномалия, характеризующаяся остановкой работы генов, или по-научному это явление вызывается отсутствием экспрессирования, что означает передачу наследственной информации от ДНК к РНК. Функция генов в данном случае заключается во включении в работу именно в свое время. Например, у мальчиков отсутствует рост волос на лице в детстве, и начинается это явление после наступления пубертатного периода. Данное заболевание впервые описали в 1956 году.
Синдром Прадера-Вилли встречается с частотой один случай на 12 000 рожденных малышей. Эту болезнь описали в своих работах Хайнрих Вилли, Андреа Прадер, Эндрю Зиглер, Алексис Лабхарт, Гвидо Фанцони.
Синдром Прадера-Вилли причины
Данное заболевание характеризуется отсутствием или не экспрессированием семи генов из пятнадцатой хромосомы, унаследованной от отца. Синдром Прадера-Вилли отмечается при изменениях в отцовской хромосоме, а в случае изменения материнской хромосомы наблюдается синдром Ангельмана.
В генном наборе, вызывающем возникновение синдрома Прадера-Вилли, копия гена, полученная от отца, активно функционирует, а материнская нет. Это значит, что когда большинство людей содержат одну рабочую копию данных генов, то больные с синдромом Прадера-Вилли живут без такой копии.
Синдром Прадера-Вилли симптомы
Для данного заболевания характерна дисплазия тазобедренных суставов, гипотонус (пониженный мышечный тонус), ожирение (предпосылки к перееданию возникают к 2-м годам). Страдающие синдромом Прадера-Вилли имеют пониженную координацию движений, а также низкую плотность костей, являются обладателями маленьких стоп и кистей, низкого роста, склонны ко сну, косоглазию, имеют искривление позвоночника. Для таких людей свойственна густая слюна, гипогонадизм (понижение функций половых желёз), наличие плохих зубов, бесплодие. Больные характеризуются задержкой психического, а также речевого развития, отставанием полового созревания и испытывают сложности в овладении навыками моторики.
Синдром Прадера-Вилли и признаки этого заболевания визуально отмечаются большой переносицей, узким и высоким лбом, миндалевидными глазами, узкими губами. Все эти признаки и симптомы не проявляются на одном больном. Зачастую у пациентов встречается до пяти вышеуказанных признаков.
Синдром Прадера-Вилли диагностика
Данное заболевание можно диагностировать еще до рождения. Об этом говорит низкая подвижность плода или зачастую неправильное положение ребенка, а также многоводие (амниотическая жидкость в чрезмерном количестве).
Синдром Прадера-Вилли зачастую диагностируется после генетического анализа. Этот анализ делают новорождённым с гипотонусом (пониженным мышечным тонусом). Иногда врачи ошибаются и диагностируют синдром Дауна или миопатию. Признаки диагностирования заболевания после рождения включают: рождение через кесарево сечение, ягодичное предлежание плода, гипотония, летаргия, слабый сосательный рефлекс по причине ослабленного мышечного тонуса малыша, трудности с дыханием, гипогонадизм.
Дети с синдромом Прадера-Вилли похожи между собой, поэтому опытный генетик сразу диагностирует болезнь, даже не дожидаясь результатов исследования крови.
Синдром Прадера-Вилли и синдром Ангельмана
На сегодняшний день известна еще одна болезнь, которую относят к сестринскому заболеванию синдрома Прадера Вилли. Эта болезнь получила название Ангельмана. Для нее свойственна мутация в материнском генетическом материале. Результаты большинства независимых исследований говорят о таких причинах возникновения этой болезни, как мутация в гене. Продуктом этого гена является ферментный компонент всей сложной системы деградации белков. Это заболевание названо именем педиатра из Британии Гарри Ангельмана, описавшего его в 1965 году.
Синдром Прадера-Вилли лечение
Заболевание, являясь врождённой генетической аномалией, остается до конца не изученным и способы его лечения так и не разработаны.
Как же лечить синдром Прадера-Вилли? Все же существуют некоторые лечебные мероприятия, позволяющие повысить качество жизни людей. К примеру, дети с гипотонусом нуждаются в массаже, а также других видах специальной терапии. Показано использование специальных методик, позволяющих улучшить развитие ребёнка, а также занятия с дефектологом и логопедом. Детям 7-ми, 8-ми, 9-ти лет с синдромом Прадера-Вилли назначаются гормоны роста, также показана гормональная терапия с использованием гонадотропинов.
Синдром Прадера-Вилли у мальчиков проявляется в гипогонадизме, микропении, а также крипторхизме (не опущении яичек). В такой ситуации врачи рекомендуют или ждать, пока яички самостоятельно опустятся, или рекомендуют хирургическое вмешательство с гормонотерапией. Чтобы откорректировать повышение веса назначается диета, ограничивающая количество углеводов и жиров. Если ожирение уже имеется, то следует отслеживать количество, а также качество пищи, которое поглощается больным с синдромом Прадера-Вилли. Для таких людей свойственен волчий аппетит. Возможно осложнение течения заболевания апноэ, характеризующееся задержкой дыхания во сне.
Синдром Прадера-Вилли прогноз
При планировании второго ребенка у этих самых родителей возможен повторный риск возникновения данного заболевания. Это зависит от механизма, вызывающего генетический сбой. Риск составляет меньше 1 % в тех случаях, когда первый ребёнок имеет делецию гена или однородительскую партеногенетическую дисомию. Риск составляет до 50 %, при условии, если сбой вызван мутацией. Риск до 25 % остается при транслокации родительских хромосом. В любом случае родителям необходимо сделать генетическое обследование.
Синдром Прадера-Вилли и его прогноз зачастую сохраняет задержку речевого, а также психического развития. Исследования, проведенные Фрим и Керфс, показали, что 5 % больных имеют коэффициент интеллекта, соответствующий среднему уровню. Это приравнивается к 85-ти и более баллов по шкале IQ. 27 % больных имеет уровень интеллекта на грани среднего, который исчисляется 70-85 баллами. 34 % больных имеет уровень интеллекта соответствующий 50-70 баллам. 27 % больных пребывает на уровне среднего отставания и исчисляются 35-70 баллами. 5 % больных имеют сильное отставание и набирают 20-35 баллов, а менее 1 % имеют значительное отставание. Другие исследования имеют данные, что 40 % больных показывают средний или сниженный интеллект.
Обычно дети с синдромом Прадера-Вилли обладают хорошей долговременной и зрительной памятью. Они способны научиться читать, обладают пассивным, богатым словарём, однако их собственная речь зачастую хуже, чем ее понимание. Также страдает и не соответствует возрастным стандартам слуховая память, навыки письма и математические навыки, кратковременная слуховая и зрительная память.
Синдром Прадера-Вилли зачастую сопровождается повышенным аппетитом, поскольку у заболевших повышается в крови уровень гормона грелина. Для больных типична пониженная концентрация соматолиберина. Одни связывают это с 15-ой хромосомой, имеющей связь с гипоталамусом. Другие данные свидетельствуют о снижении общего числа клеток, а также окситоцин содержащих клеток в паравентрикулярных ядрах гипоталамуса. Но что интересно имеются данные, которые говорят об обратном: вскрытие умерших с этим заболеванием зачастую не показывает дефектов гипоталамуса.
vlanamed.com
Синдром Прадера-Вилли
Синдром Прадера-Вилли относится к категории наследственных заболеваний, при которых отмечается целый комплекс аномалий, развивающихся вследствие значительного повреждения генетического материала.
История изучения
Впервые клинические признаки данной патологии были описаны в середине прошлого столетия Генрихом Вили и Андреа Прадером, в честь этих клиницистов и был назван данный синдром.
Эпидемиология
Синдром Прадера-Вилли в популяции встречается с частотой 1 случай на 10-25 тысяч новорожденных. На сегодняшний день имеются описания 400 тысяч случаев данной патологии.
Генетический механизм, ставший причиной развития данного расстройства, определяет уровень риска рождения в семье с больным ребёнком ещё одного больного потомка. Униотцовская дисомия или делеция гена снижают неблагоприятный прогноз до 1%, хромосомная транслокация предполагает вероятность рождения ещё одного больного ребёнка на уровне 25%, а проблемы, связанные с импритингом, могут в 50% случаев стать причиной повторного появления дефекта генетического материала у потомков пары.
Этиология
Клинические проявления, имеющие место при данной патологии, формируются вследствие распространённого повреждения генетического материала, причём в данном случае речь идёт о хромосоме, унаследованной от отца, а именно пятнадцатой, на которой удалены или не реализуют в полной мере свою функцию семь генов или несколько из них.
У детей с данной патологией материнская копия указанных генов не функционирует, в результате чего не происходит восстановление генетического материала, утраченного вследствие патологических процессов, затрагивающих отцовскую пятнадцатую хромосому.
Клиническая картина
Все клинические проявления данной патологии условно можно разделить на несколько групп в зависимости от времени их возникновения. Ещё в период внутриутробного развития о наличии генетической аномалии свидетельствует необычное положение плода в матке, беспричинное снижение его двигательной активности, многоводие.
Часто дети с данной генетической аномалией имеют ягодичное предлежание в анамнезе. Сразу же после рождения ребёнка обращают на себя внимание такие симптомы, как выраженная гипотония новорожденного, слабый сосательный рефлекс, затруднения, возникающие при дыхании, недоразвитие наружных половых органов.
В раннем детстве на первый план выступает задержка физического и умственного развития ребёнка, чрезмерная сонливость и повышенная утомляемость, сколиоз, сформировавшийся ещё внутриутробно, косоглазие.
В дальнейшем диагностируются задержка речевого развития, слишком быстрый набор веса вследствие регулярного переедания, нарушение формулы сна, проблемы с координацией. В подростковом возрасте отмечается позднее половое созревание, чрезмерная гибкость, низкий рост, ожирение значительной степени.
Дети сталкиваются с серьёзными трудностями при обучении, так как в большинстве случаев отмечаются различные нарушения интеллекта, при этом визуальное восприятие информации таких детей может быть на высоком уровне, а последовательная и слуховая обработка различной информации часто затруднены, краткосрочная слуховая и зрительная память не соответствуют возрастным нормам.
Дети с данной патологией испытывают серьёзные затруднения при написании текстов и выполнении математических действий. К наиболее часто диагностируемым при данном патологическом состоянии поведенческим расстройствам относятся гиперфагия, ведущая к ожирению, повышенная тревожность и частые подёргивания.
Только у 5% детей с данным синдромом отмечаются патологические психиатрические симптомы в виде депрессии, паранойи и галлюцинаций.
Диагностика
Внешний вид ребёнка (низкий рост, маленькие конечности, лёгкая умственная отсталость и гипогонадизм) позволяет заподозрить наличие генетических аномалий. Установить степень и размеры генетического дефекта даёт возможность генетическое тестирование.
Лечение
Специфическое лечение для данной нозологии не разработано. При постановке диагноза на ранних стадиях заболевания применяется адекватная заместительная гормональная терапия, позволяющая обеспечить нормальный линейный рост и становление половой функции.
Низкий мышечный тонус является показанием к проведению лечебной физкультуры и физиотерапевтических процедур, отклонения в интеллектуальной сфере, нарушения речи требуют занятий с профессионалами.
Прогноз
Чем раньше был установлен диагноз и начато симптоматическое лечение, тем более благоприятен прогноз для жизни и здоровья ребёнка.
Вы можете посмотреть комментарии или написать свой.
kotikit.ru
Синдром Прадера-Вилли: описание, диагностика, фото, лечение
Синдром Прадера-Вилли считается очень редким генетическим нарушением, при котором семь генов, находящихся в 15-й отцовской хромосоме, полностью либо частично отсутствуют и не функционируют в нормальном режиме.
Данная генетическая патология возникает по причине того, что правильно функционирует только полученная от отца копия определенного гена. В копии от матери также наблюдаются некоторые нарушения. Рассмотрим подробнее.
В организме здорового человека присутствуют копии генов, благодаря которым органы могут функционировать без каких-либо отклонений от нормы. При развитии синдрома Прадера-Вилли подобные копии отсутствуют. В настоящее время известны заболевания, которые по своей сути похожи на эту болезнь.
Похожий механизм возникновения может наблюдаться также при синдроме Ангельмана, но и при этом мутации поражают генетический материал, полученный от матери. Подобные заболевания, как правило, проявляют себя в различных формах и имеют различную степень тяжести. Но, тем не менее, они неизлечимы.

Причины развития синдрома
Синдром Прадера-Вилли - наследственная детерминированная патология, которая развивается лишь при развитии некоторых аномалий. Другими словами, при определенных хромосомных нарушениях начинают страдать родительские гены, что приводит к серьезным изменениям. Клиническая картина развивается, когда семь генов в 15-й отцовской отсутствуют либо не экспрессируются. При этом заложенная информация в ДНК не преобразуется в РНК.
Ученые, которые занимались выяснением причин возникновения данной наследственной патологии, ранее считали, что из-за такого отклонения образуется гомозигота. Затем сделали вывод, что преобладающие признаки присутствуют в аутосомах, и главным путем передачи болезни является наследование.
Генетики проводили многочисленные цитогенетические анализы патологии, с помощью которых было установлено, что у отцов детей, пораженных болезнью, произошла транслокация 15 хромосомы. Фото детей с синдромом Прадера-Вилли представлены в нашей статье.

Генетический механизм
На сегодняшний день точно установлено, что при данной патологии 15-я хромосома повреждается в сегментах от q11.2 до q13. То же самое происходит при синдроме Ангельмана. Однако данное заболевание характеризуется совершенно иными симптомами. Подобный диссонанс могут объяснить лишь только таким явлением в генетической науке, как геномный импринтинг, а также унипарентальная дисомия.
При унипарентальной дисомии обе хромосомы наследуются лишь от одного родителя, но для того чтобы это произошло, на генный материал должны влиять определенные биохимические факторы. Этот факт установлен при помощи прометафазных анализов и ДНК-маркирования некоторых локусов данной хромосомы.
Синдром Прадера-Вилли обусловлен двумя основными механизмами: микроделецией 15-й хромосомы, полученной от отца, и идиосомией материнских хромосом, обе из которых получены от матери.
При геномном импринтинге изменения фенотипа зависят от того, в чьих хромосомах – отца или матери - произошла экспрессия.
Синдром Прадера-Вилли у детей
Механизмы нарушений, происходящих в организме больного синдромом, еще не изучены до конца. Однако при этом у них присутствует ряд присущих только этому виду заболеваний симптомов. Считается, что пациенты набирают вес из-за усиления формирования жировых клеток и понижения уровня липолиза.
Кроме того, имеют место дисфункции гипоталамуса, которые преимущественно отмечаются в двух его ядрах – вентролатеральном и вентромедиальном. Подобные процессы приводят к сбоям в формировании вторичных половых признаков. Сниженная активность тироназы в фолликулах волос и меланоцитах приводит к тому, что волосы, кожа и радужка глаза гипопигментируются.
Какие же основные симптомы синдрома Прадера-Вилли?

Симптомы заболевания
Данная патология может обнаружиться еще на начальных сроках беременности при неправильном расположении плода и при его малой подвижности. Кроме того, у беременной может существенно меняться уровень гонадотропина, вырабатываемого клетками хориона, и присутствовать симптомы многоводия. На основании этих симптомов нельзя поставить диагноз, тем не менее, они могут быть достаточным основанием для дальнейшей диагностики.
Дисплазия
У детей синдром Прадера-Вилли (на фото выше) может выражаться в наличии врожденных вывихов бедра (дисплазии), в ослаблении мышечного тонуса, а также в нарушениях координации. Известны случаи, когда новорожденный не мог самостоятельно сосать и проглатывать грудное молоко. При этом нарушении питание осуществляется при помощи зонда. Могут возникать нарушения дыхания, и в некоторых случаях требуется искусственная вентиляция органов дыхания.
Сонливость
Помимо этого, существуют и другие симптомы болезни Прадера-Вилли. Например, у детей может наблюдаться повышение сонливости. Что же касается внешних факторов, то у ребенка наблюдаются задержки в развитии. Поэтому для подобных больных характерны низкий рост, недоразвитые кисти и стопы, часто развивается косоглазие.
Другие симптомы
В дальнейшем данная патология характеризуется следующими симптомами:
- Искривление позвоночного столба.
- Кариес молочных зубов и повышение густоты слюны.
- Склонность к перееданиям.
- Гипофункции половых желез, которые в дальнейшем приводят к бесплодию.
- Ожирение.
- Задержка моторики и речевого развития.
- Отсталость в психомоторном развитии.
- Запоздалость в половом созревании.
Данные симптомы определяются визуально. В подростковый период выявляются следующие симптомы:
- Задержка речевых навыков.
- Избыточный вес при очень низком росте.
- Неестественная гибкость тела.
- Снижение интеллекта и неспособность к обучению.

Диагностика синдрома Прадера-Вилли
Данную наследственную патологию можно заметить еще во время внутриутробного развития при проведении УЗИ. В подобных случаях женщинам рекомендуются определенные виды пренатальной диагностики, а при необходимости специалисты применяют инвазивных способы решения проблемы.
После родов опытный специалист вправе поставить диагноз «Болезнь Прадера-Вилли» уже на первоначальном осмотре младенца. Тем не менее, чтобы подтвердить его, необходимы специальные генетические тестирования. Также в крови матери исследуется содержание хорионического гонадотропина. Благодаря таким методам есть возможность выявить субмикроскопические и функциональные патологии на ДНК-уровне.

По каким критериям ставится диагноз?
Диагноз может быть поставлен по следующим клиническим критериям:
- При рождении низкий вес и рост ребенка в случаях доношенной беременности.
- Неправильное положение, в том числе ягодичное предлежание плода.
- Другие микроаномалии в развитии.
- Выраженная гипотония мышечной системы.
- Сниженная пигментация кожи и волос.
- Ожирение, развивающееся, как правило, к шести месяцам.
- Задержки психологического, моторного и речевого развития.
Дети, у которых наблюдается данный синдром, постоянно требуют еды и очень мало двигаются. Из-за чрезмерного набора массы тела у них может наблюдаться такое осложнение, как апноэ, что часто является причиной смерти во сне.
В чем состоит лечение синдрома Прадера-Вилли?
Лечение
До настоящего момента не существует конкретных методов лечения синдрома. Терапия при этом является, как правило, симптоматической. Если у новорожденного определяются проблемы с дыхательной деятельностью, то его переводят на искусственную вентиляцию легких, а при проблемах с глотанием ставят желудочный зонд, посредством которого проводится энтеральное питание. В случаях снижения тонуса мышц показан массаж и разнообразные физиотрапевтические методы.
Детям с болезнью Прадера-Вилли ежедневно вводится рекомбинантный гормон роста, который поддерживает увеличение массы мышц и помогает уменьшать аппетит больного. Также осуществляется замещение хорионического гонадотропина.
Во время подобного заболевания наблюдается гипогонадизм, то есть недостаточная развитость половых желез и изменение функций половой системы. В этом случае осуществляется заместительная гормональная терапия, позволяющая стимулировать рост и половое созревание.
В некоторых случаях детям с задержками речи и недостаточностью психологического развития может потребоваться помощь психиатра или психолога. И главное – необходимо постоянно контролировать объем пищи, которую они потребляют. Детям с синдромом Прадера-Вилли назначается специальная диетотерапия.
Риск того, что второй ребенок семейной пары, у которых первый страдает данным заболеванием, родится с такими же генетическими проблемами, невероятно высок. Родителям в подобном случае рекомендуется пройти консультацию, где специалисты всестороннее обследуют их и подсчитают риски.
Дети с болезнью Прадера-Вилли нуждаются в постоянном наблюдении эндокринолога и невролога.

Улучшение общего самочувствия на фоне заболевания
Среди людей, у которых имеется синдром, значительно повышены показатели соматической заболеваемости, затруднено общение, возникает потребность в специфической помощи, обусловленной особенностями их заболевания. Они могут не понимать, зачем необходимо заботиться о своем здоровье. Если состояние удовлетворительно и больной чувствует себя хорошо, качество жизни его улучшается.
Необходимо устранить следующие факторы:
- Повышенный риск внезапной смерти.
- Вероятность заболеваения.
- Увеличение количества факторов, которые определяют материальное благополучие.
- Недостаточный доступ к оздоровительным услугам и медицинскому обслуживанию.
Люди с патологией Прадера-Вилли имеют особые потребности, обусловленные их основным заболеванием. Они нуждаются в особой терапии острых и хронических патологий, в помощи в укреплении общего здоровья и т. д. Их потребности должны быть удовлетворяться в специальных учреждениях, обеспечивающих медицинскую помощь, которая, в свою очередь, может состоять в лечении основного заболевания и соматических нарушений, связанных с основной патологией.
Какова продолжительность жизни с синдромом Прадера-Вилли? Данное заболевание часто приводит к снижению длительности жизни больных до 60 лет. Однако прогноз на выздоровление таких людей весьма неутешительный.
В статье было представлено подробное описание синдрома Прадера-Вилли. Теперь вы знаете, что это за патология.
загрузка...
worldfb.ru
Синдром Прадера-Вилли: описание, диагностика, фото, лечение
Синдром Прадера-Вилли считается очень редким генетическим нарушением, при котором семь генов, находящихся в 15-й отцовской хромосоме, полностью либо частично отсутствуют и не функционируют в нормальном режиме.
Данная генетическая патология возникает по причине того, что правильно функционирует только полученная от отца копия определенного гена. В копии от матери также наблюдаются некоторые нарушения. Рассмотрим подробнее.
В организме здорового человека присутствуют копии генов, благодаря которым органы могут функционировать без каких-либо отклонений от нормы. При развитии синдрома Прадера-Вилли подобные копии отсутствуют. В настоящее время известны заболевания, которые по своей сути похожи на эту болезнь.
Похожий механизм возникновения может наблюдаться также при синдроме Ангельмана, но и при этом мутации поражают генетический материал, полученный от матери. Подобные заболевания, как правило, проявляют себя в различных формах и имеют различную степень тяжести. Но, тем не менее, они неизлечимы.

Причины развития синдрома
Синдром Прадера-Вилли - наследственная детерминированная патология, которая развивается лишь при развитии некоторых аномалий. Другими словами, при определенных хромосомных нарушениях начинают страдать родительские гены, что приводит к серьезным изменениям. Клиническая картина развивается, когда семь генов в 15-й отцовской отсутствуют либо не экспрессируются. При этом заложенная информация в ДНК не преобразуется в РНК.
Ученые, которые занимались выяснением причин возникновения данной наследственной патологии, ранее считали, что из-за такого отклонения образуется гомозигота. Затем сделали вывод, что преобладающие признаки присутствуют в аутосомах, и главным путем передачи болезни является наследование.
Генетики проводили многочисленные цитогенетические анализы патологии, с помощью которых было установлено, что у отцов детей, пораженных болезнью, произошла транслокация 15 хромосомы. Фото детей с синдромом Прадера-Вилли представлены в нашей статье.

Генетический механизм
На сегодняшний день точно установлено, что при данной патологии 15-я хромосома повреждается в сегментах от q11.2 до q13. То же самое происходит при синдроме Ангельмана. Однако данное заболевание характеризуется совершенно иными симптомами. Подобный диссонанс могут объяснить лишь только таким явлением в генетической науке, как геномный импринтинг, а также унипарентальная дисомия.
При унипарентальной дисомии обе хромосомы наследуются лишь от одного родителя, но для того чтобы это произошло, на генный материал должны влиять определенные биохимические факторы. Этот факт установлен при помощи прометафазных анализов и ДНК-маркирования некоторых локусов данной хромосомы.
Синдром Прадера-Вилли обусловлен двумя основными механизмами: микроделецией 15-й хромосомы, полученной от отца, и идиосомией материнских хромосом, обе из которых получены от матери.
При геномном импринтинге изменения фенотипа зависят от того, в чьих хромосомах – отца или матери - произошла экспрессия.
Синдром Прадера-Вилли у детей
Механизмы нарушений, происходящих в организме больного синдромом, еще не изучены до конца. Однако при этом у них присутствует ряд присущих только этому виду заболеваний симптомов. Считается, что пациенты набирают вес из-за усиления формирования жировых клеток и понижения уровня липолиза.
Кроме того, имеют место дисфункции гипоталамуса, которые преимущественно отмечаются в двух его ядрах – вентролатеральном и вентромедиальном. Подобные процессы приводят к сбоям в формировании вторичных половых признаков. Сниженная активность тироназы в фолликулах волос и меланоцитах приводит к тому, что волосы, кожа и радужка глаза гипопигментируются.
Какие же основные симптомы синдрома Прадера-Вилли?

Симптомы заболевания
Данная патология может обнаружиться еще на начальных сроках беременности при неправильном расположении плода и при его малой подвижности. Кроме того, у беременной может существенно меняться уровень гонадотропина, вырабатываемого клетками хориона, и присутствовать симптомы многоводия. На основании этих симптомов нельзя поставить диагноз, тем не менее, они могут быть достаточным основанием для дальнейшей диагностики.
Дисплазия
У детей синдром Прадера-Вилли (на фото выше) может выражаться в наличии врожденных вывихов бедра (дисплазии), в ослаблении мышечного тонуса, а также в нарушениях координации. Известны случаи, когда новорожденный не мог самостоятельно сосать и проглатывать грудное молоко. При этом нарушении питание осуществляется при помощи зонда. Могут возникать нарушения дыхания, и в некоторых случаях требуется искусственная вентиляция органов дыхания.
Сонливость
Помимо этого, существуют и другие симптомы болезни Прадера-Вилли. Например, у детей может наблюдаться повышение сонливости. Что же касается внешних факторов, то у ребенка наблюдаются задержки в развитии. Поэтому для подобных больных характерны низкий рост, недоразвитые кисти и стопы, часто развивается косоглазие.
Другие симптомы
В дальнейшем данная патология характеризуется следующими симптомами:
- Искривление позвоночного столба.
- Кариес молочных зубов и повышение густоты слюны.
- Склонность к перееданиям.
- Гипофункции половых желез, которые в дальнейшем приводят к бесплодию.
- Ожирение.
- Задержка моторики и речевого развития.
- Отсталость в психомоторном развитии.
- Запоздалость в половом созревании.
Данные симптомы определяются визуально. В подростковый период выявляются следующие симптомы:
- Задержка речевых навыков.
- Избыточный вес при очень низком росте.
- Неестественная гибкость тела.
- Снижение интеллекта и неспособность к обучению.

Диагностика синдрома Прадера-Вилли
Данную наследственную патологию можно заметить еще во время внутриутробного развития при проведении УЗИ. В подобных случаях женщинам рекомендуются определенные виды пренатальной диагностики, а при необходимости специалисты применяют инвазивных способы решения проблемы.
После родов опытный специалист вправе поставить диагноз «Болезнь Прадера-Вилли» уже на первоначальном осмотре младенца. Тем не менее, чтобы подтвердить его, необходимы специальные генетические тестирования. Также в крови матери исследуется содержание хорионического гонадотропина. Благодаря таким методам есть возможность выявить субмикроскопические и функциональные патологии на ДНК-уровне.

По каким критериям ставится диагноз?
Диагноз может быть поставлен по следующим клиническим критериям:
- При рождении низкий вес и рост ребенка в случаях доношенной беременности.
- Неправильное положение, в том числе ягодичное предлежание плода.
- Другие микроаномалии в развитии.
- Выраженная гипотония мышечной системы.
- Сниженная пигментация кожи и волос.
- Ожирение, развивающееся, как правило, к шести месяцам.
- Задержки психологического, моторного и речевого развития.
Дети, у которых наблюдается данный синдром, постоянно требуют еды и очень мало двигаются. Из-за чрезмерного набора массы тела у них может наблюдаться такое осложнение, как апноэ, что часто является причиной смерти во сне.
В чем состоит лечение синдрома Прадера-Вилли?
Лечение
До настоящего момента не существует конкретных методов лечения синдрома. Терапия при этом является, как правило, симптоматической. Если у новорожденного определяются проблемы с дыхательной деятельностью, то его переводят на искусственную вентиляцию легких, а при проблемах с глотанием ставят желудочный зонд, посредством которого проводится энтеральное питание. В случаях снижения тонуса мышц показан массаж и разнообразные физиотрапевтические методы.
Детям с болезнью Прадера-Вилли ежедневно вводится рекомбинантный гормон роста, который поддерживает увеличение массы мышц и помогает уменьшать аппетит больного. Также осуществляется замещение хорионического гонадотропина.
Во время подобного заболевания наблюдается гипогонадизм, то есть недостаточная развитость половых желез и изменение функций половой системы. В этом случае осуществляется заместительная гормональная терапия, позволяющая стимулировать рост и половое созревание.
В некоторых случаях детям с задержками речи и недостаточностью психологического развития может потребоваться помощь психиатра или психолога. И главное – необходимо постоянно контролировать объем пищи, которую они потребляют. Детям с синдромом Прадера-Вилли назначается специальная диетотерапия.
Риск того, что второй ребенок семейной пары, у которых первый страдает данным заболеванием, родится с такими же генетическими проблемами, невероятно высок. Родителям в подобном случае рекомендуется пройти консультацию, где специалисты всестороннее обследуют их и подсчитают риски.
Дети с болезнью Прадера-Вилли нуждаются в постоянном наблюдении эндокринолога и невролога.

Улучшение общего самочувствия на фоне заболевания
Среди людей, у которых имеется синдром, значительно повышены показатели соматической заболеваемости, затруднено общение, возникает потребность в специфической помощи, обусловленной особенностями их заболевания. Они могут не понимать, зачем необходимо заботиться о своем здоровье. Если состояние удовлетворительно и больной чувствует себя хорошо, качество жизни его улучшается.
Необходимо устранить следующие факторы:
- Повышенный риск внезапной смерти.
- Вероятность заболеваения.
- Увеличение количества факторов, которые определяют материальное благополучие.
- Недостаточный доступ к оздоровительным услугам и медицинскому обслуживанию.
Люди с патологией Прадера-Вилли имеют особые потребности, обусловленные их основным заболеванием. Они нуждаются в особой терапии острых и хронических патологий, в помощи в укреплении общего здоровья и т. д. Их потребности должны быть удовлетворяться в специальных учреждениях, обеспечивающих медицинскую помощь, которая, в свою очередь, может состоять в лечении основного заболевания и соматических нарушений, связанных с основной патологией.
Какова продолжительность жизни с синдромом Прадера-Вилли? Данное заболевание часто приводит к снижению длительности жизни больных до 60 лет. Однако прогноз на выздоровление таких людей весьма неутешительный.
В статье было представлено подробное описание синдрома Прадера-Вилли. Теперь вы знаете, что это за патология.
загрузка...
buk-journal.ru